COT/COM/COC Annual Report 2021
About the Committees
In this guide
In this guide
From 2021 onwards Annex 6: Index to Subjects and Substances considered in previous annual reports has now become its own web page: Index | Committee on Toxicity.
PDF documents on this page are not in a fully accessible format, if you require it to be fully accessible, please see the HTML page for that document.
This is the 31st joint annual report of the Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT), the Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment (COM) and the Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment (COC).
The aim of these reports is to provide a brief background to the Committees' decisions. Those seeking further information on a particular subject can obtain details from the Committee’s statements and minutes, available from the websites listed below or from the Committee’s administrative Secretary.
In common with other independent advisory committees, Committee members are required to follow a Code of Conduct which also gives guidance on how commercial interests should be declared. Members are required to declare any commercial interests on appointment and, again during meetings if a topic arises in which they have an interest. If a member declares a specific interest in a topic under discussion, and it is considered to be a conflict of interest, he or she may, at the Chairman's discretion be allowed to take part in the discussion but is excluded from decision-making. Annex 1 contains the terms of reference under which the Committees were set up. The Code of Conduct is at Annex 2 and Annex 3 describes the Committees’ policy on openness.
Annex 4 is the Good Practice Agreement for Scientific Advisory Committees. Annex 5 contains a glossary of technical terms used in the text. Annex 6 is an alphabetical index to subjects and substances considered in previous reports. Previous publications of the Committees are listed at Annex 7.
These three Committees also provide expert advice to other advisory committees, such as the Scientific Advisory Committee on Nutrition, and there are links with the FSA Science Council, Veterinary Products Committee and the Expert Committee on Pesticides (formerly the Advisory Committee on Pesticides).
The Committees’ procedures for openness include the publication of agendas, finalised minutes, agreed conclusions and statements. These are published on the internet at the following links:
This report contains summaries of the discussions and links to the Committees’ published statements. Paper copies are available upon request to the Secretariats.
Committee on the Toxicity of Chemicals in Food, Consumer Products and the Environment - Preface 2021
In this guide
In this guide
Head and shoulders Image of Prof Alan Boobis, standing in front of a patterned background. Prof Boobis is wearing half framed glasses and a light-coloured striped shirt.
It is 30 years since the COT issued its first report, also joint with COC and COM. In that time a lot has changed, but not the core function of the Committee, which remains to provide advice on the safety-in-use and on the potential adverse effects of chemicals in food, whether added intentionally or present incidentally.
At the beginning of year, Dr Sarah Judge, Newcastle University, became vice-chair of the Committee, for which I would like to thank her very much.
The Committee met on seven occasions during 2021, undertaking a busy and varied programme of work. The continuing COVID pandemic meant that the COT again held its meetings virtually but was able to successfully adapt to this new way of working to function effectively over the year. However, we look forward to being able to meet in person again as soon as that becomes possible.
The Committee has commenced a review of contaminants and other chemicals in support of the risk assessment of the maternal diet now being undertaken by the Scientific Advisory Committee on Nutrition (SACN). A number of topic areas were considered including environmental contaminants, excess nutrients and food supplements and the priority compounds identified; reviews of vitamin D, iodine and ginger were then started. The Committee also continued to work on another ongoing programme of work, on biologically based food contact materials - considering chitosan and bamboo composites as part of this.
Other topics discussed by the Committee this year have covered a wide range including variable lifetime exposure to chemicals, the combined effects of mycotoxins, , biomonitoring, oral exposure to microplastics, and the final EFSA opinion on titanium dioxide. Several previously reviews, including electronic nicotine delivery systems (e-cigarettes) and novel heat-not-burn tobacco products, have been updated along with cannabidiol where information on non-oral exposure was added to the position paper on CBD.
The Committee also discussed a roadmap setting out the way towards achieving the regulatory acceptance of New Approach Methodologies. These new techniques, including in silico modelling and in vitro assays, provide an important opportunity to not only reduce the use of laboratory animals but also have the potential to provide approaches that are faster, cheaper and more tailored in risk assessment. This was followed up in a virtual workshop which took place in October 2021. The FSA and COT are taking a UK lead on this important area.
The Committee also contributed comments to a number of public consultations from EFSA including on non-monotonic dose response and a draft protocol for the assessment of phthalates.
COT and COC Members along with other experts have been collaborating in a Working Group examining the Synthesis of Epidemiological and Toxicological Evidence (SETE). The resulting report was published in the Spring of 2021 and is an excellent example of the really valuable work that can be done by collaboration between the different Scientific Advisory Committees.
A joint Working Group has been set up between the COT and SACN colleagues to undertake a benefit- risk assessment of plant-based drinks consumed as an alternative to cows’ milk. It is hoped this will report in 2022.
This year, the Committee said goodbye to Professor Faith Williams. On behalf of all Members, I would like to express the COT’s sincere thanks to her for all her invaluable contributions to the work of the Committee over the years.
We welcomed new Members Professor Shirley Price from the University of Surrey, Professor Thorhallur Ingi Halldorsson from the University of Iceland and Dr Simon Wilkinson from Newcastle University to the Committee and look forward to working with them.
Next year it is expected that the work of the Committee will begin to change as it starts to oversee and assure the risk assessment of regulated products, which were previously assessed in Europe. To that end, three Joint Expert Groups (JEGs) have been established as part of the FSA Scientific Advisory Committee (SAC) structure and these JEGs will advise the FSA on regulated products; along with other SACs, the COT will oversee the work of these Groups and the Committee looks forward to working with them.
I would like to thank my fellow Committee Members for their commitment and invaluable contributions to the work of the Committee in very challenging circumstances. I would also like to express my sincere appreciation to the Secretariat who, despite the many difficulties they faced with the virtual meeting format and an evolving regulatory environment, continued to provide first class support for the Committee.
Professor
Alan Boobis (Chair)
OBE PhD CBiol FRSB FBTS FBPhS
COT Evaluations 2021
In this guide
In this guideThe potential risk(s) of combined exposure to mycotoxins
1.1 The Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT) has identified the potential risk(s) from combined exposure to mycotoxins as a possible concern during their review of mycotoxins in the diet of infants and young children.
1.2 Mycotoxins are secondary metabolites produced by plant fungi under particular climate and biological conditions and can cause adverse health effects in both humans and animals. Those of greatest concern to human health are produced by several groups of filamentous fungi, namely Aspergillus, Fusarium and Penicillium species.
1.3 Mycotoxins are stable, low-molecular weight chemicals and are often not affected by food processing (e.g., cooking).
1.4 Cereals (e.g. wheat, oats, rice, corn (maize), barley, sorghum, rye, and millet) are often the crops most severely affected; however, some nuts, fruits and spices can also be affected.
1.5 Advances in analytical techniques have allowed the simultaneous detection and quantification of multiple mycotoxins in both food and animal feed.
1.6 Climate change could have a significant impact on mycotoxin production. Changes in the climate are expected to affect levels of rainfall, humidity, temperature etc., which in turn, influence mycotoxin production, which varies for each individual pathogen species and/or strain.
1.7 Current government and industry regulations are usually based on assessing the risks from individual mycotoxins and, at most, group metabolites with the parent compound, but take no account of the varied dynamics and potential interactions between co-occurring groups of mycotoxins.
1.8 In light of this, new combinations of factors (mycotoxins/host plants and geographical location) will need to be considered when assessing the potential risk(s) from dietary exposure to mycotoxins.
1.9 Based on the available information, the COT was unable to complete a risk assessment on the potential risk(s) from combined exposure to mycotoxins for several reasons. These include:
- A lack of harmonisation of approaches/methodologies and data analysis/modelling for toxicological investigations.
- The underlying mechanisms of interactions between individual mycotoxins in different combination(s) have yet to be fully understood.
- There is little information on the potential toxic effect(s) of mycotoxin mixtures on the gut microbiota.
1.10 Considerations for possible co-exposures from breastmilk and weaning foods also need to be considered for infants and young children.
1.11 Co-occurrence data in food is scarce, and the available methods for multi-mycotoxin detection in food samples are still not harmonised for use in a regulatory setting. In addition to this the following need further consideration for a robust exposure assessment:
- The management data for which the true values are below the limit of detection and could not be accurately determined.
- The consistent and well-defined use of probabilistic models and methodologies for multi-biomarker studies that estimate levels of exposure to multiple mycotoxins in biological samples (e.g. urine).
1.12 The COT noted that there was a lack of UK specific data, particularly in biomonitoring; however, there were a number of studies ongoing and additional information will be available in the future. The Public Health England Secretariat informed COT Members that the UK will not be collecting new data for mycotoxins under the Human Biomonitoring for the European Union Initiative; however, in the future, more data could be obtained through Health Protection Research Units. The results of such research would be of potential value in the risk assessment of co-exposures to mycotoxins.
1.13 COT Members recommended that as a pragmatic first step, a review should be carried out of the mycotoxins that appeared to show a common effect on protein synthesis (i.e., DNA or RNA synthesis), assuming dose additivity, and that frequently co-occur in food commodities – an exposure estimate could be performed and the estimates compared with the recommended health-based guidance values to calculate the Margin of Exposure or the Hazard Index utilised, to determine whether there is any potential concern from co-exposure to these mycotoxins in UK consumers.
1.14 Depending on the outcome of this screening risk assessment, research may be needed on those mycotoxins affecting ribosomal protein synthesis, to determine whether they do in fact exhibit dose additivity in their effects, to help develop a reliable basis for their cumulative risk assessment.
The full COT statement, including references, can be found on the COT website: Statement on the potential risk(s) of combined exposure to mycotoxins 2021.
Overarching statement on the potential risks from exposure to microplastics
1.15 As part of horizon scanning, the Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT) identified the potential risks from microplastics as a topic it should consider. Upon review of the literature, it was decided that nanoplastics should also be included. An initial scoping paper was presented to the COT in October 2019 (TOX/2019/62). Since then, the topic and additional information has been discussed several times by COT with the final substantive discussion in December 2020.
1.15 The purpose of this overarching statement is to bring together these discussions, summarise the COT conclusions reached to date and provide a high-level overview of the current state of knowledge, data gaps and research needs with regards to this topic.
1.16 Future sub-statements, which will consider in detail the potential toxicological risks of exposure to microplastics via the oral and inhalation routes, are intended to provide supplementary material for this overarching statement. The Committee will review the potential risks from oral exposure of microplastics (resulting from their presence in food and bottled drinks). A review of the potential risks of microplastics via the inhalation route will be produced jointly with the Committee of Medical Effects of Air Pollutants (COMEAP) Secretariat at Public Health England. The need for additional reviews of other significant routes of exposure will also be considered.
1.17 Micro- and nanoplastics are widespread. They are either intentionally added to products or occur as a result of plastics being fragmented down into smaller sizes by natural processes such as wear, weathering and corrosion. There is no internationally agreed definition of what a microplastic is, however, the most widely used size range is 0.1 to 5,000 µm. Plastic particles that are smaller than the lower range are considered nanoplastics (i.e. 1 nm to 0.1 µm).
1.18 The COT noted that there is little data on the effects of microplastics on mammals (including humans) whether taken in orally or via inhalation. Some microplastics are excreted from the body (~>90%) but small amounts of others may remain in the gut (gastrointestinal tract; GIT) or move from the GIT into organs or tissues (via endocytosis by M cells and paracellular persorption). No epidemiological or controlled dose studies that evaluated the effects of orally ingested microplastics in humans were identified. There is a similar lack of information on inhaled microplastics.
1.15 As such, the COT concludes that based on the available data, it is not yet possible to perform a complete assessment for the potential risks from exposure to micro and nanoplastics via the oral and inhalation routes. However, the Committee concurs with the conclusions reached by other authoritative bodies (EFSA, 2016; WHO, 2019; SAPEA, 2019; SAM, 2020; ECCC and HC, 2020) that further research is required to better identify target tissues, threshold doses, and the toxic mode(s) of action for any toxicity observed.
1.17 The COT concluded that the literature data on exposure to particles from tyre wear would need separate consideration from microplastic exposure from food, since the particles were chemically quite different (in their polymeric nature). Risk assessment of such material was considered potentially outside the scope of the current exercise.
1.18 The most significant data gaps are the lack of appropriate and harmonised analytical methods for the detection of micro- and nanoplastics (together with suitable reference standards), as well as information on their toxicokinetic and toxicity profiles in/relevant to humans.
1.19 The COT highlighted that additional information will be needed from all exposure sources, which include indoor and outdoor air, dust and soil, before a risk assessment can be completed. The presence of MPs in food and water needs to be put into perspective with other sources of MPs such as atmospheric fallout.
1.20 Comprehensive assessment of microplastics and contaminant concentrations in different foods and the impact of cooking (on the release of and subsequent bioavailability of contaminants/leachates) need to be further investigated to better understand the implications for human health.
1.21 Current studies typically focus only on one type of particle/tissue interaction. As such, further research is necessary to explore the effects of the range of particle types in different tissues in silico, in vitro and in vivo. The range of particle types studied should also take account of emerging/novel plastic-based materials such as bioplastics.
The full COT statement, including references, can be found on the COT website: Microplastics Overarching Statement 2021.Page Break
Sub-statement on the potential risk(s) from exposure to microplastics: Oral route
1.22 The purpose of this sub-statement is to provide supplementary material to the overarching statement (COT Statement 2021/02) and to consider in detail the potential toxicological risks of exposure from microplastics ingested via the oral route (i.e. resulting from the presence of microplastics in food, drinking water and bottled drinks).
1.23 The COT noted that there are limited data regarding the toxicokinetic fate of orally ingested microplastics in mammalian species, and that microplastic particles can either translocate from the gastrointestinal tract (GIT) into organs or tissues (via endocytosis by M cells and paracellular persorption), and/or be excreted. The extent to which retention in the mammalian GIT tract is of concern, if at all, is not yet clear. No epidemiological or controlled dose studies in which the effects of orally ingested microplastics in humans have been evaluated were identified.
1.24 As such, the COT concludes that based on the available data, it is not yet possible to perform a complete assessment for the potential risks from exposure to micro and nanoplastics to humans via the oral route. It should be noted that the COT’s conclusions are consistent with those reached by other authoritative bodies, as described in the COT overarching statement on the potential risks from exposure to microplastics; COT Statement 2021/02; please refer to paragraphs 101-129).
1.25 The COT previously considered the extent to which exposure to tyre wear (a source of synthetic polymeric material) might contribute to the total burden of adverse effects of nano- and microplastics (NMPs) in humans (Annex B of TOX/2020/15). The COT concluded, however, that the literature data on exposure to particles from tyre wear would need separate consideration from microplastic exposure from food, since the particles were chemically quite different in their polymeric nature. Risk assessment of such material was considered to be outside the scope of the current exercise.
1.26 The most significant data gaps are the lack of appropriate and harmonised analytical methods for the detection and characterisation of micro- and nanoplastics (together with suitable reference standards), as well as information on their toxicokinetic and toxicity profiles in/relevant for humans.
1.27 The COT highlighted that additional information will be needed on all exposure sources, which include indoor and outdoor air, dust and soil before a holistic risk assessment can be completed. The presence of MPs in (sea)food and water needs to be put into perspective with other sources of MPs such as atmospheric fallout.
1.28 Comprehensive assessment of microplastics and contaminant concentrations in different foods and the impact of cooking on the desorption and subsequent bioavailability of contaminants/leachates, need to be further investigated to better understand the implications for human health.
1.29 Current studies typically focus on only one type of particle/tissue interaction, as such, further research is necessary to explore the effects of the range of particle types in different tissues in vitro and/or in vivo. These range of particle types should also take account of emerging/novel plastic-based materials such as bioplastics.
The full COT sub-statement can be found on the COT website: Sub-statement on the potential risk(s) from exposure to microplastics: Oral route 2021.
Consumption of plant-based drinks in children aged 6 months to 5 years of age
Introduction
1.30 The Department of Health and Social Care (DHSC), Public Health England (PHE) and the Food Standards Agency (FSA) are receiving an increasing number of enquiries regarding the use of plant-based drinks in the diets of infants and young children. Therefore, the COT was asked to consider the potential risks posed by soya, almond and oat drinks consumed in the diets of these age groups.
1.31 The UK government advises that first infant formula (which is usually based on cows’ milk) is the only suitable alternative to breast milk in the first 12 months of a baby’s life. Whole cows’ milk can be given as a main drink from the age of 1 year. From this age, unsweetened calcium-fortified plant-based drinks, such as soya, almond and oat drinks can also be given to children, as part of a healthy, balanced diet.
1.32 The main challenge in the assessment of the safety of these drinks is the lack of information regarding dietary intakes for infants and young children following dairy-free or plant-based diets.
1.33 Organisations providing recommendations for ensuring a balanced diet for vegan children under 5 were used to identify appropriate portion sizes and consumption frequency to develop representative intake scenarios for children following dairy-free or plant-based diets. These were then used to calculate daily intake figures for different age groups in order to calculate exposure to the chemicals of concern in the different drinks.
1.34 Although the exposure estimates made the best use of the available data, there was a high degree of uncertainty with regards to actual intakes. This was because these figures were based on recommendations to ensure that dietary requirements for infants and children of these ages were met. Actual intakes may be different.
1.35 The Committee agreed to use the previously adopted approach of assuming that a child’s consumption was exclusively of a single plant-based drink as it is possible that young children may develop a preference for one drink. This was regarded as the most cautious approach because it assumes the highest intakes.
1.36 The need for real-world consumption information for people following plant-based diets in all age groups was highlighted by the Committee, as the popularity of these diets is increasing and information on realistic dietary intakes would help inform future risk assessments.
Soya
1.37 Soya drinks are a popular alternative to dairy products and their use is becoming more widespread. Soya products contain phytoestrogens (in the form of isoflavones). Concerns about adverse effects from isoflavones in the diet of infants and young children relate principally to their ability to mimic the female hormone, oestrogen, and therefore their potential impact on development and reproduction.
1.38 The safety of phytoestrogens was considered by the COT in 2003 and 2013. In 2003, the Scientific Advisory Committee on Nutrition (SACN) considered the COT outputs and concluded that there was no scientific basis for changing the current government advice – namely, that there is no substantive medical need for, nor health benefit arising from the use of soya-based infant formula, and that it should be used only in exceptional circumstances to ensure adequate nutrition, such as for babies who have cows' milk allergy. In 2013 this was reconfirmed by the COT. Currently, soya formula should be used only if it has been recommended or prescribed by a health visitor or GP.
1.39 For this evaluation, the Committee reviewed data published since the 2013 evaluation. The Committee concluded that new animal studies did not add significantly to the overall database.
1.40 As with previous evaluations, although there was some indication of possible adverse effects in human studies, it was not possible to determine from the available data, whether sensitivity to phytoestrogens varies among different age groups.
1.41 The Committee concluded that the intakes of phytoestrogens from consumption of soya drinks in children aged 6 months to five years was no greater than the estimated maximum intake by infants aged 0 – 6 months consuming soya formula where medically necessary (see paragraph 9 above). This maximum level of phytoestrogen intake was estimated to be 9.5 mg/kg bw per day.
1.42 The Committee agreed that, based on the available information, exposure to phytoestrogens from other soya-based products in the diets of children aged 6 months to 5 years of age was lower than that from soya drinks, and therefore of less concern. It was, however, noted that when exposure to phytoestrogens from all sources of soya in the diet was considered, the exposure came much closer to the maximum level of 9.5 mg/kg bw per day.
1.43 Members agreed that, in addition to potential toxicological concerns, consideration of nutritional issues would also be required to assess whether it was necessary to issue additional advice on the consumption of soya-based drinks in children aged 6 months to 5 years of age.
Oats
1.44 Oat drinks can be given to children following plant based or dairy- free diets, as an alternative to cows’ milk. Oats can be contaminated with mycotoxins, notably the trichothecene mycotoxins T-2 and HT-2, deoxynivalenol (DON), and Ochratoxin A (OTA). Mycotoxins are naturally occurring toxins produced by certain moulds. As such, they are unavoidable contaminants in certain foods, like oats. International standards are in place to limit exposures to mycotoxins to the lowest possible levels. The COT evaluated the available data and considered the estimated exposures to the above contaminants.
T2 and HT-2
1.45 The European Food Safety Authority (EFSA) considered the safety of T-2 and HT-2 in 2017. Health-based guidance values were established for emetic effects (causing vomiting) following acute (short term or single) exposure, and for immune- and hepatotoxicity effects (toxic effects on the liver) following long-term exposure. After reviewing UK intake data, COT concluded that in terms of acute exposure to the sum of HT-2 and T-2, consumption of a large quantity of oat drink (minimum of 5.4L/ day) was required to exceed the Acute Reference Doses (ARfD). Thus, acute exposure to HT-2 & T-2 from the consumption of oat drink was considered to be of low risk.
1.46 Generally, all long term exposures for T-2, HT-2 were below the respective TDI, with the exception of minor exceedances observed in children aged 1-2 years old for T-2 and HT-2. The assessment of total exposure from oat drinks combined with the general diet was considered conservative (i.e., high compared with likely reality) and as the exceedances were minor and transient in nature, it was concluded that there would be no chronic health effects in respect to T-2 and HT-2.
DON
1.47 For DON, a group Tolerable Daily Intake (TDI) was established for the sum of DON, and its related compounds, 3-Ac-DON, 15-Ac-DON and DON-3-glucoside based on animal studies in which body weight gain was reduced. Vomiting was identified as the critical effect following acute exposure in humans.
1.48 COT concluded that in terms of acute exposure to DON, consumption of a large quantity of oat drink (minimum 28L/d) was required to exceed the Acute Reference Dose (ARfD). Thus, acute exposure to DON was considered to be of low risk.
1.49 Generally, all long term exposures for T-2 and HT-2 were below the TDI, with the exception of minor exceedances observed in children aged 1-5 years old. The assessment of total exposure from oat drinks combined with that from the general diet was considered conservative and as the exceedances were minor and transient in nature, it was concluded that there would be no chronic health effects in respect to DON.
OTA
1.50 For OTA, EFSA in 2020 established a Margin of Exposure (MOE) approach for neoplastic and non- neoplastic effects (kidney tumours and microscopic kidney lesions, respectively) to assess the risk posed by OTA. The MOE is a measure that is used to determine the level of exposure at which there starts to be a safety concern. For genotoxic carcinogens, MOEs ≥10, 000 indicate low concern. For other effects, an MOE ≥100 indicates low concern. It is not clear whether OTA can cause kidney tumours by directly interacting with the DNA (genotoxic carcinogen), or via a different mechanism.
1.51 It was noted that there were many uncertainties in the cancer endpoint used for risk characterisation, and furthermore, it was unclear whether or not OTA was a genotoxic carcinogen and thus which MOE threshold value would be applicable. The Committee noted that the MOE of ≥10,000 for substances that are directly genotoxic and carcinogenic may not be appropriate in this case because there is some evidence that OTA does not interact directly with DNA. Some age groups had MOEs lower than desirable for non-neoplastic changes while all age groups had MOEs lower than 10,000 for cancer effects. The uncertainty in the assessment was considered to be high, especially considering the lack of analytical information on the presence of these contaminants in oat drinks and the assumptions used in the exposure assessment. It was noted that it is likely that the risk was being overestimated.
1.52 In respect of OTA, the Committee was unable to conclude whether the exposure estimates indicated a potential health concern. It was agreed that assessments of actual exposure are needed for adults as well as young children, to establish whether there were potential health concerns for the general population.
1.53 Overall, it was concluded that for the sum of DON and T-2 and HT-2, based on the available data there was no risk to health. However due to the uncertainties in the available dataset, the risk from exposure to OTA could not be determined.
Almonds
1.55 Almond drinks have a lower nutritional value than soya or oat drinks, however they can be given to children as an alternative to cows’ milk. The mycotoxin, aflatoxin B1 was identified as a possible chemical contaminant in almonds, which could be potentially transferred to almond drinks. Aflatoxin B1 is a genotoxic carcinogen, so the EU sets a legal limit for the amount of aflatoxin which can be present; this is called the maximum level and uses the ‘as low as reasonably achievable’ (ALARA) principle. This is to ensure that exposure to such compounds is at the lowest possible level. As no more reliable data on aflatoxin levels were available, it was assumed that the almonds contained aflatoxin at the legal maximum level.
1.56 The lack of analytical information on the effect that processing of almonds during almond drink manufacture has on the levels of aflatoxins, as well as the lack of information on the levels in almond drinks themselves, was considered the main limitation in assessing the risk to health. Considering the above limitations, it was concluded that undertaking a risk assessment based on the Maximum Levels set by EFSA was highly uncertain and was likely to lead to an overestimation of risk and therefore was not appropriate. The risk to health from exposure to AFB1 could not be determined.
1.57 Almonds also contain cyanogenic glycosides, which can be released when the almond is physically broken down by chewing or processing. When this happens, they may interact with the enzyme ß-glucosidase, also present in almonds. This enzyme breaks down the cyanogenic glycosides and can yield hydrogen cyanide. Exposure to large amounts of hydrogen cyanide can lead to convulsions, loss of consciousness, dizziness, weakness, mental confusion and heart failure.
1.58 High levels of glycosides are present in bitter almond varieties, whereas there is very little present in sweet varieties. The quantity of cyanogenic glycosides present in almond drinks is uncertain, but only low levels of cyanide have been detected on analysis. Available information indicates that bitter almond varieties are not grown in commercial almond orchards and although the inadvertent use of bitter almonds in almond milk drinks cannot be completely ruled out, bitter almonds would not be deliberately used as they would be unpalatable, imparting a strong ‘marzipan’ flavour to the drink. Overall, Members agreed that there were no specific concerns for acute toxicity from cyanogenic compounds in almond drinks.
Position paper on the alternatives to conventional plastics for food & drinks packaging
1.59 In conjunction with pressure from environmentally aware consumers and the strategy to reach net zero to mitigate the effects of climate change recent years have seen a major global increase in the development and use of alternative biobased materials to conventional plastics for food and drinks packaging.
1.60 These alternatives are a diverse, complex set of materials and blends. The materials are usually derived from living matter (animal, plant or fungal biomass) and are partially or wholly made of substances that are naturally available or are synthesised from biomass, such as sugarcane, corn, and algae. Some examples include, but are not limited to, wheat straws; beeswax wraps to replace clingfilm; and bamboo/rice husk for paper coffee cups.
1.61 The alternative materials are usually classified into three main groups: bio-based plastics, biodegradable plastics and compostable materials.
Advice on biobased food contact materials (BBFCMs) has been increasingly requested from the Food Standards Agency (FSA) so it was therefore considered timely for the Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT) to review the available toxicological information on BBFCMs.
1.62 Several papers have been presented to the COT, which included discussion of the following topics: the limited research that has been undertaken into the development of BBFCMs and the associated potential risks to the consumer; relevant market data and reports; a table of enquiries received from the FSA Food Contact Material (FCM) Policy Team - these included Non-intentionally added substances (NIAS) such as the presence of formaldehyde in bamboo cups and the allergic potential of material such as chitin and wheat; as well as a detailed discussion paper focussing on the immunogenicity and allergenicity of chitin and chitosan-based BBFCMs.
1.63 The COT acknowledged the challenges and complexities associated with BBFCMs as well as highlighting several limitations and knowledge gaps on BBFCMs research and regulation. These included labelling, composition (including biodegradability), contaminants and standardisation. Members noted that quantitative information was needed on contamination, degradation, migration of chemicals and allergens during the manufacture and use of commercial BBFCMs, as well as environmental impacts after disposal, such as the formation of micro/nanoparticles upon entering landfill or from energy-from-waste processes. It was noted that only limited evidence exists to demonstrate BBCFMs in direct food-contact applications meet similar standards of safety as conventional plastics.
1.64 Members agreed that there was a general lack of information on the presence of nanomaterials in BBFCMs. Therefore, overall, information on specific migration of all the possible migrating substances (nanofillers, plasticizers, antimicrobial additives, micron and nano sized plastic particles etc.) under different testing conditions would improve identification of potential hazards and enable an estimation of possible exposure. This would allow better demonstration that these novel biodegradable packaging materials meet comparable requirements. Additional toxicity studies or approaches to enable assessment of long term risk may be needed for a more comprehensive risk assessment.
1.65 The COT agreed a priority list of BBFCMs for health risk assessment based on their potential health hazards, extent of usage, and UK policy interest. The prioritised materials to be reviewed are: polylactic acid (PLA), starches, bamboo biocomposites and polyhydroxyalkanoates (PHA). This was not a closed list, other priority BBFCMs could be added as necessary based on the same criteria.
Health risk assessments of the prioritised BBFCMs should be considered within the context of life cycle assessment studies, which include environmental hazards to address indirect impacts on human health. However, this was not all within the remit of the COT. It was noted that the Department for Environment, Food and Rural Affairs (DEFRA) (and its expert scientific committee, the Hazardous Substances Advisory Committee, HSAC), the Organisation for Economic Cooperation and Development (OECD), and the Environment Agency were assessing the wider environmental impacts. These impacts should be monitored to identify additional potential hazards to human health.
1.66 Further assessments of intelligent packaging (also known as smart packaging) and nanomaterials used within food packaging will be undertaken as policy priorities and resources permit as part of the Committee’s work and would include bio sensors as well as nano coatings.
The full COT statement can be found on the COT website: Position paper on the alternatives to conventional plastics for food & drinks packaging.
Review of the EFSA opinion on dioxins
1.67 The COT reviewed the scientific basis and implications for risk management of the new EFSA tolerable weekly intake (TWI) for dioxins and considered that there were substantial uncertainties over the derivation of the TWI and possible inconsistencies between the animal and human data. Given the implications for risk management, the Committee felt that the rationales for the choices of key studies were not sufficiently clear in the published opinion, which made it difficult to evaluate the strength of the evidence. These concerns meant that the COT was unable to endorse the opinion and considered it necessary to reconsider the evidence base and set its own tolerable intake.
1.68 EFSA established a new TWI of 2 pg/TEQ/kg bw, which is 7-fold lower than its previous tolerable intake, based on data from a Russian Children’s study, identifying semen quality, following pre- and postnatal exposure, as the critical effect. The COT noted this study appeared inconsistent with the findings in a second study and considered the Russian study to provide only a weak data set. The studies on experimental animals (rodents) included in the EFSA evaluation confirmed that developmental effects occurred at body burdens similar to those used as the basis for the previous risk assessment. However, the COT considered there were inconsistencies in the animal data presented in the EFSA opinion and was unclear, in particular, regarding the rationale for the selection of the study to evaluate the critical body burdens. The COT had raised specific concerns about their reliability in 2001 and later FSA commissioned studies to address these concerns, which failed to replicate the specific findings but found other reproductive effects at similar body burdens. Overall, the data presented in EFSA’s opinion implied that humans were more sensitive to dioxins than rats. However, this would be inconsistent with the existing body of data on dioxins and knowledge on the relative sensitivity of the human and rat aryl hydrocarbon receptor (AHR). Due to these uncertainties, the COT did not agree with the newly established TWI and the 7-fold reduction in the TWI appeared too conservative for the database overall. The Committee was unable to comment on the dietary exposures and whether they should be compared to the new TWI.
1.69 The European Commission (EC) has not yet adopted EFSA’s new TWI due to ongoing work at the international level to review the basis and values of the WHO toxic equivalent factors (TEFs). The review of the TEFs and a finalised assessment by the EC are not expected until 2022, at the earliest. The COT noted that this also presupposes that the effects of concern are mediated via the AHR.
1.70 The Committee acknowledged that a further review of dioxins would be an extensive and lengthy undertaking. However, even if the current HBGV were immediately reduced, it would take decades to reduce body burden in the population, due to the nature of dioxins, especially their long half-life in humans. The current COT TDI was based on the most sensitive endpoint in the animal studies and is intended to protect the most sensitive population group, hence it would also be protective for all population groups and for other less sensitive effects.
1.71 Thus, while the re-assessment of dioxins is a necessary and important piece of work going forward, the COT did not consider it necessary in the meantime to alter its existing advice on dioxins. The COT considered that their current TDI of 2 pg/kg bw per day is protective for effects on the developing male fetus, that this was supported by later studies on this endpoint and was consistent with their consideration of the WHO-TEF concept.
COT principles for assessing risks from less than lifetime exposure or variable exposure over a lifetime
1.73 Dietary exposures to chemicals are typically compared to a health-based guidance value (HBGV), for example a tolerable daily intake (TDI), that has been established to be safe for long term exposure. Such values set a level of exposure that is considered acceptable if continued throughout a normal lifetime, i.e., it is an upper amount to which an individual can be exposed daily over a lifetime without a significant risk to health.
1.74 Sometimes people may be exposed to chemicals at a higher level for a shorter period of time. The COT produced a statement containing COT recommendations on possible ways of refining the risk assessment for such less-than-lifetime exposures. The statement includes a flowchart to illustrate the process, which is reproduced in Figure 1, below.
The full COT statement can be found on the COT website: Statement on COT principles for assessing risks from less than lifetime exposure or variable exposure over a lifetime.
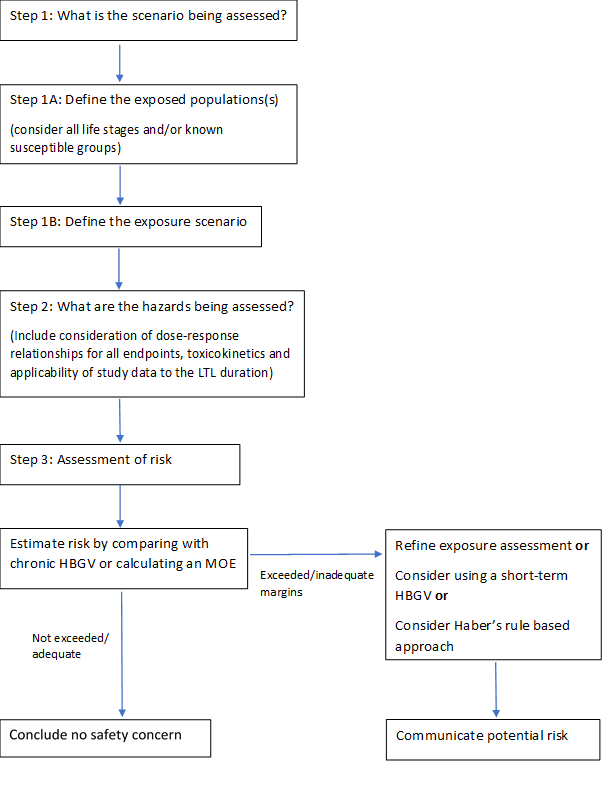
This is a flowchart setting out the steps needed to assess variable or less than lifetime exposure to a chemical.
Figure 1: Flowchart to illustrate the process of assessing risks from less than lifetime or variable (LTLV) exposures. Where appropriate, toxicokinetic or toxicodynamic modelling could be applied to refine any of the steps.
Development of Human Biomonitoring Guidance Values in the HBM4EU project
1.75 The Committee were asked to comment on the methodology for the derivation of human biomonitoring guidance values by the European Human Biomonitoring Initiative, referred to as HBM4EU, which is a project designed to develop a harmonised and systematic strategy for the derivation of human biomonitoring guidance values (HBM-GVs).
1.76 Members considered other types of human biomonitoring guidance values to allow comparison with established methods and discussed the potential application of the HBM4EU strategy and values, as well as their relevance to the UK.
1.77 There were two aspects that needed to be considered: the generation of the human biomonitoring guidance values and the application of these values to the population. It was also noted that, similar to determining any guidance value, the derivation of the human biomonitoring guidance values would depend on the type of data available and on establishing the relationship between the exposure and the effect. UK specific biomonitoring data would be useful for risk assessment and more information (such as appropriate auxiliary data) would be required before being able to use these values for this purpose.
1.78 In terms of the methodology for deriving the human biomonitoring guidance values, the values would need to be validated from a toxicological perspective. Ideally, exposure could be correlated to environmental levels in combination with human biomonitoring data, for example by collaborating with the agencies such as the Environment Agency or Defra to collect environmental biomonitoring exposure data. Correlation of National Diet and Nutrition Survey (NDNS) data with environmental biomonitoring data would be useful to refine exposures.
1.79 There may be insufficient toxicological data to establish human biomonitoring guidance values and a continuation project with targeted studies to allow for the generation of suitable data may be necessary.
1.80 On occasion, both external and internal guidance values will be needed - for example in cases where there is variability in the exposure depending on the product, and therefore monitoring of both product levels and internal levels in humans would be needed; this would need to be done on a case by case basis. Human biomonitoring guidance values are not often used stand alone, but they add value when they can be used in combination with other approaches
1.81 Further information would be useful on the pharmacokinetic requirements needed to establish a biomonitoring equivalent and it was noted that the sampling and exposure scenarios needed to fit sampling time. Requirements for marker substances were not included in the paper. Appropriate data on dermal exposure would also be important in ensuring the assumptions made were correct.
1.81 The Committee agreed that the strategy developed by HBM4EU was robust and scientifically valid, depending on kinetics information and data availability. In principle, the use of HBM-GVs derived by the HBM4EU in the UK would be possible. In practice, and in line with any other guidance value, detailed evaluation of the human biomonitoring value would be needed to determine whether the critical endpoint was appropriate for the UK population.
1.82 Going forward, the use of human biomonitoring guidance values in risk assessment could be helpful to the FSA and the Committee was content to review future case studies and offer their perspective. However, if endorsement of these values was needed, the Committee would have to perform a detailed evaluation to offer their perspective.
1.83 This topic has also been discussed by the COC (see paragraph 3.1 below)
First draft non-technical statement on how the Committees evaluate the relevance and reliability of data when assessing a chemical of concern
1.84 This topic was brought to the COT by the COC Secretariat.
1.85 Guidance aimed at a lay audience had been prepared, providing clarity on how the expert committees evaluate data with respect to consideration of biological relevance and statistical significance.
1.86 The topic arose during COC horizon scanning activities and the draft guidance for a number of years. the draft guidance been revised following review by lay members of the COC, COT and COM.
1.87 The COT considered the guidance was largely appropriate for the purpose of describing the mechanisms of ascribing biological and statistical significance to the assessment of the risk posed to the consumer by a chemical, but acknowledged that the statistical methods described were potentially overly complex for a lay readership. However, any simplification of the definition of concepts, such as the null hypothesis and p-value, should ensure that their meaning was lost.
1.88 The Committee noted that information on the workings of the sister committees should be included on the Committee website. However, further information was needed on some aspects, for example, how a particular chemical or issue was added to the agenda, how the risks to the consumer from it were assessed, and the basis of the conclusions reached. However, some of these aspects are covered in the Committee Code of Practice, albeit briefly.
1.89 The Committee made a number of additional minor suggestions for amendments.
Review of EFSA Scientific opinion on the safety assessment of titanium dioxide as a food additive (E171)
1.90 The COT was asked to comment on the “Scientific opinion on the safety assessment of titanium dioxide as a food additive (E171) “ published by EFSA in May of 2021. In this opinion, the EFSA panel concluded that on the basis of the currently available evidence along with all the uncertainties, in particular the fact that the concern regarding genotoxicity could not be resolved, that E171 can no longer be considered as safe when used as a food additive.
1.91 The EFSA Opinion had also been presented to the COM for comments (see paragraph 2.33).
1.92 The Committee note the COM’s preliminary comments, regarding the quality of the data and the difficulties in evaluating it adequately from the description given in the opinion. The lack of a good dataset and a well-defined test compound (due to the poorly defined specifications) are also considered as severe limitations. The COM consider the mechanism of genotoxicity appears to be indirect and probably has a threshold and, that the positive effects observed in the genotoxicity studies could be attributed to the nano-fraction of titanium dioxide.
1.93 The COT agree with the COM view and note the large discrepancy between the underlying dataset and the conclusions drawn by EFSA. On the genotoxicity of nanoparticles, it was noted that this could either be a concentration effect leading to oxidative damage or a stress effect, however, it was unclear as the results in different cell lines were equivocal and inconsistent. It was also noted that in some tests titanium dioxide had shown less reactivity.
1.94 In several parts of the Opinion, published papers are presented at face value, and there is no discussion of the results nor the Weight of Evidence to support the conclusions being made. There are also discrepancies and conflicts between the results of the studies reported and the overall conclusions.
1.95 On balance, the Committee considers that the weight of evidence does not support the conclusions drawn by EFSA. The Committee also agree with the comments of the COM with regards to risk communication that “As it stands the conclusion is highly risk adverse based on the weak evidence available, and it might create unnecessary concern to the public.” Care should be taken when expressing such conclusions in a binary manner given the extensive uncertainties in the dataset.
1.96 The COT suggested that the COM should independently review the database on genotoxicity and apply the COM’s Guidance on determining thresholds.
1.97 EFSA’s concluded that no differentiation could be made with regards to size/form of titanium dioxide and different aspects of toxicity, however, it seems likely that nanoparticles may be driving the toxicity.
1.98 It was decided that an interim position paper, capturing the COT’s view and the proposed next steps should be published. This can be found at: COT position paper on titanium dioxide
Updated COT Evaluations 2021
In this guide
In this guideCannabidiol (CBD)
Updated CBD position paper- Position paper on the potential risk of CBD in CBD food products: additional text summarising Committee discussions relating to dermal and inhalation exposure.
1.84 The COT ‘Position paper on the potential risk of CBD in CBD food products’ published in July 2020 summarised the discussions and conclusions of the COT and COM from July 2019 to May 2020 on the available toxicological information of relevance to cannabidiol (CBD) in non-medicinal food products.
Dermal exposure to CBD
1.85 The Committee discussed data of relevance to dermal exposure to CBD from CBD-containing cosmetics products. Such products include serums, creams, washes/rinse-off products, bath products, deodorants, balms, and toothpastes.
1.86 Dermal exposure to CBD may contribute to systemic exposure and/or local effects. Although absorption levels would probably be low because the compound is lipophilic, repeat application could lead to accumulation in the stratum corneum and subsequent slow diffusion into the systemic circulation. Overall, the Committee considered that dermal absorption of CBD was unlikely to be greater than from oral exposure and may be lower. Dermal absorption of CBD was likely to be less than 10% compared with oral absorption. The Committee noted that absorption of CBD from cosmetic products may also occur via inhalation of sprays and mists generated during product use. Dermal pharmaceutical CBD products may differ from cosmetic CBD products, as these may have formulations designed to maximise dermal absorption.
1.87 There was insufficient information on the pharmacokinetics and toxicity of dermal CBD to conduct a risk assessment of the safety of CBD in cosmetic products.
1.88 No conclusions could be drawn on whether dermally applied CBD poses a safety concern, nor on the potential for drug interactions. The risk from aggregate exposure to multiple CBD products, including cosmetics, could not be determined due to lack of information. No good quality in vivo or in vitro data were available to allow estimation of systemic doses.
1.89 Overall, the Committee noted that additional exposure through topically applied CBD could potentially occur, and this would increase overall systemic exposure of CBD. However, there are data gaps that need to be addressed to be able to evaluate the potential for adverse effects related to dermal exposure to CBD.
Exposure to CBD by inhalation
1.90 Inhalation exposure to CBD may occur via various sources, for example smoking CBD-containing plant material, use of electronic nicotine (and non-nicotine) delivery systems (E(N)NDS) containing e-liquids to which CBD has been added, or from aerosolised therapeutic applications.
1.91 The nature of the source material will affect the risk assessment, for example in terms of the presence or absence of thermal degradation products, and because different delivery methods may affect the bioavailability of CBD.
1.92 The available evidence base relating to potential adverse effects of inhaled CBD is small. However, some conclusions on the likelihood of toxicity from the inhalation of CBD can be inferred based on oral data. Inhalation exposures pose a potential safety concern and adverse effects could be greater than those from an equivalent oral dose as the bioavailability of inhaled CBD is often higher compared with oral exposure. Following absorption across the lung, the type of adverse effects occurring would be independent of route of exposure. Inhibitory drug interactions would be expected at levels comparable to those following oral exposure, given the apparent higher bioavailability across the lung compared with the gut. Effects on the central nervous system would be expected following inhalation, thus a health warning might be necessary relating to driving or using heavy machinery.
1.93 Some experimental data suggest a possible interaction of CBD with steroids could be a cause for concern, however this is an area of research that is currently not well understood.
1.94 Overall, there was insufficient information to generate a risk assessment regarding the safety of use of CBD in products intended for inhalation, but the available data indicated caution. The Committee agreed that the recommended upper limit of 1 mg/kg body weight per day established for dietary exposure to CBD should be applied to total combined exposure, including that from inhalation.
1.95 As a result of the COT discussions, some additional text was added to the existing position paper which summarises the discussions around dermal and inhalation exposure for inclusion in an updated position paper.
The full updated COT position paper can be found on the COT website: Updated position paper on the potential risk of CBD in CBD food
Statement on the potential toxicological risks from electronic nicotine (and non-nicotine) delivery systems (E(N)NDS – e cigarettes): presence and pharmacokinetics of nicotine salts
1.96 At the end of 2020 and in 2021, the Committee considered data on the presence and pharmacokinetics of nicotine salts in electronic nicotine delivery system (ENDS) products.
1.97 It was agreed that this should be included as an addendum to the COT statement on the potential toxicological risks from electronic nicotine (and non-nicotine) delivery systems (E(N)NDS – e-cigarettes).
The addendum to the statement will be published in due course.
Committee Procedures
In this guide
In this guideDraft EFSA Scientific Committee Opinion on scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals
1.84 In May 2021, EFSA released draft guidance, prepared by its Scientific Committee, on the grouping of chemicals for risk assessments of combined exposure to multiple chemicals. The Committee were asked to comment on the draft opinion as part of EFSA’s public consultation process.
1.85 Overall, the Committee agreed that the proposed guidance provides a pragmatic and scientifically sound approach for grouping chemicals for a combined risk assessment.
1.86 The main comments of the Committee were as follows:
· Sorting different chemicals into assessment groups on the basis of common key events is appropriate but for data-poor chemicals, this may result in the formation of very large chemical assessment groups (CAGs), particularly if grouping is done on the basis of adverse effects, such as potential liver effects.
- Although the scientific criteria for dose addition were provided in the draft EFSA guidance, the underlying assumption of dose addition is not clearly stated.
- With regards to the prioritisation methods for grouping chemicals into assessment groups, the default threshold values appeared to be rather arbitrary, and not entirely supported by scientific data; thus, the threshold values should be tested, and re-evaluated after some time.
- Appendix C (‘statistical methods to study the probability of combined risk or combined exposure’) was not directly referred to in the draft guidance document. It would be useful to have some examples where these statistical methods were used, such as use of correlation matrices for multivariate pattern analysis. Furthermore, it may be possible to obtain a high probability of co-exposure (‘r’ value) from assessment of a low number of chemicals.
Draft EFSA Scientific Committee Opinion on biological plausibility of non-monotonic dose responses and their impact on the risk assessment
1.87 In 2016, the European Food Safety Authority (EFSA) published the results of a contracted-out report on a systematic review of the existing literature where signs of non-monotonic dose responses (NMDRs) had been observed (Beausoleil et al., 2016). In the report, the scientific evidence for such NMDRs was assessed with a systematic review being performed in line with the EFSA guidance. The report extracted dose-response datasets from studies having at least 5 dose groups, which were then analysed by the PROAST software package. The strength of the evidence was characterised using visual/statistics-based checkpoints.
1.88 The EFSA Scientific Committee (SC) was asked to prepare a scientific opinion on the biological relevance, if any, of the apparent non-monotonic dose responses identified in the commissioned report and to address the possible consequences for the human health risk assessments conducted by EFSA. The COT was asked to comment on the opinion as part of the public consultation process. The opinion is a review of the previous methods used for assessing the presence of non-monotonic dose responses, not of the responses.
1.89 The COT made a number of specific comments which are presented below:
- A critical review of the key studies claiming NMDR is needed to compare against, for example, OECD guidelines, and to more fully address randomisation.
- Some of the evidence supporting the study showing a biphasic effect on heart rate was not included, suggesting that the conclusion regarding NMDR, or otherwise, could be seen as biased.
- Consideration was not given as to whether NMDR might affect the upper and lower confidence limits of the Benchmark dose (BMD), even if the curve was fitted only to those data points before the sign of the dose-response changed.
- The implications of NDMR of key events at low doses in the context of homeostatic control needs greater consideration.
- The opinion concludes that if an effect for which NMDR is observed is an apical effect and NMDR is supported by further experimental work, no further investigations are needed. The corollary of this is that when such an observation was not supported by further experimental investigations, more work was needed. This meant that the opinion only provides for two possibilities 1) a conclusion of NMDR or 2) that more work was needed.
- Ethical justification is needed for the increased animal use that would be necessary in order to have sufficient data points to fully explore non-monotonicity. Moreover, possible confounders should be taken into account, and study design reviewed carefully before committing further resources to investigating possible nonmonotonicity.
- It was unclear whether the Scientific Committee’s view is that there are additional data on apical effects suggesting that relevant NDMR do occur; and, if this is the case, then it is unclear why these were not considered in the earlier reports. Conversely, if the data suggested these effects do not occur, then it appears to be unclear why there is emphasis later on the need to consider the possible implications of NMDR at low doses, which should be investigated on a case by case basis (e.g. “in cases where biological considerations or previous results suggest that NMDR may be present”). Hence, the overall message of this opinion could be clearer.
EFSA draft opinion on “Identification and prioritisation for risk assessments of phthalates, structurally similar substances potentially used as plasticisers in materials and articles intended to come into contact with food” and “draft protocol for the exposure assessment as part of the safety assessment of phthalates, structurally similar substances potentially used as plasticisers in materials and articles intended to come into contact with food”
1.90 EFSA published a “draft opinion on identification and prioritisation for risk assessments of phthalates, structurally similar substances potentially used as plasticisers in materials and articles intended to come into contact with food” and a “draft protocol for the exposure assessment as part of the safety assessment of phthalates, structurally similar substances potentially used as plasticisers in materials and articles intended to come into contact with food” for public consultation on the 5th of November 2021.
1.91 The new assessment follows on from EFSA’s previous update on the risk assessment of five phthalic acid esters (ortho-phthalates), namely di-butylphthalate (DBP), butyl-benzyl-phthalate (BBP), bis(2- ethylhexyl)phthalate (DEHP), di-isononylphthalate (DINP) and di-isodecylphthalate (DIDP) for use in FCMs, in December 2019.
1.92 The Committee was asked to comment on the draft opinion as part of the public consultation process.
1.93 The main toxicological concern for phthalates are adverse effects on reproduction, with a mode of action involving fetal testosterone reduction. It is difficult to group phthalates for hazard assessment purposes, given that reproductive toxicity is not the main toxicological outcome for all substances (i.e., DIMP and DIPP). Oher compounds with different toxicities have yet to be assessed, including some higher molecular weight phthalates. The current EFSA prioritisation list is bases on the previous assessment date of phthalates. However, the COT some of these compounds were currently undergoing further assessment by ECHA, and hence additional data with a focus on genotoxicity and reproductive effects may be forthcoming.
1.94 Overall, the approaches proposed by EFSA to prioritise phthalates and the corresponding assessment of their exposure are logical and pragmatic. However, until a complete list and toxicological profile for these substances is available, further comment on the (hazard) assessment would prove difficult.
1.95 Clearer information on exposure assessment would be helpful. A deterministic approach can result in an overestimation of exposure while a probabilistic approach could be potentially more realistic, especially if human biomonitoring is used to validate the findings. It is a positive step that the EFSA approach appears to be integrating human biomonitoring data. However, Members further information should be provided on how PBPK modelling would be used to interpret the human biomonitoring data.
1.96 It may prove difficult to exclude and/or separate occupational exposure within biomonitoring data. Occupational data may contribute significantly to overall exposure, potentially more so than the diet. A questionnaire on occupational exposure may be beneficial to gather additional information on this.
1.97 The exposure protocol Is sensible and it is useful to include exposure in EFSA’s prioritisation process. However, until data are available and estimation of combined exposures is possible, the current approach is mostly theoretical.
1.98 EFSA will not be considering the UK population as part of their exposure assessment, hence the FSA may need to consider how to follow up on EFSA’s evaluation from a UK perspective.
Public Consultation on Code of Practice for Scientific Advisory Committees and Councils
1.128 The Code of Practice for Scientific Advisory Committees and Councils’ (CoPSAC) applies to science advisory committees and councils affiliated to the UK government that provide independent expert advice to facilitate decision making. CoPSAC has been revised based on feedback received from Committee and Council stakeholders, and a wider consultation was now taking place. The consultation was aimed at academics and other experts who provide science advice to the UK government and sought views on the independence, transparency, diversity, and inclusion aspects of the CoPSAC in particular.
1.129 The Committee made a number of comments.
- In the recruitment section, there needed to be a mention of how to increase diversity through different channels of advertisement.
- Further clarification was needed to distinguish declarations of interest and conflicts of interest.
- More clarity is required on how SAC Members are appointed.
- More information was needed on lay membership. The document implies that the appointment of lay Members is not mandatory, and there is also a need to clarify the expectations of lay Members.
- Section 5.5 concerning liability might be perceived as unintentionally negative. The penalty section needs to be revised and details on conduct need to be made clearer.
- The Committee noted section 7.1 on the environmental impact, including attendees’ travel. Whilst the environmental impacts are considered to have been lower for virtual meetings, the quality of discussions in virtual versus in-person meetings may differ. Confidentiality may need to be reviewed, as this may be harder to control in a virtual meeting. However, virtual meetings may allow for greater diversity, as they may permit access for individuals who might otherwise be unable to attend in person. For future meetings, hybrid options could be useful.
- Guidance on the retention of both digital and physical documents by Members would be helpful.
COT Ongoing Work 2021
In this guide
In this guideThe COT risk assessment of substances in the diet of women in preconception, pregnancy and up to 24 months post-partum
Background
1.130 The Scientific Advisory Committee on Nutrition (SACN) last considered maternal diet and nutrition in relation to offspring health in its 2011 reports ‘The influence of maternal, fetal and child nutrition on the development of chronic disease in later life’ and the 2018 report ‘Feeding in the first year of life”. In the latter report, the impact of breastfeeding on maternal health was also considered. In 2019, SACN agreed to conduct a risk assessment on nutrition and maternal health focusing on maternal outcomes during pregnancy, childbirth and up to 24 months after delivery; this would include the effects of chemical contaminants and excess nutrients in the diet. SACN agreed that, where appropriate, other expert Committees would be consulted and asked to complete relevant risk assessments, e.g., in the area of food safety advice. Accordingly, the COT were asked to contribute to this project.
Prioritisation of xenobiotics
1.131 Following discussion of the prioritisation papers on substances to be considered for risk assessment, the Committee agreed that some substances were of sufficient concern to be allotted individual papers and others could be grouped together into an overarching Statement.
1.132 The substances for which individual papers were requested are:
- Vitamin D, iodine, caffeine, vitamin A, ginger, ochratoxin a, fumonisins, zearalenone, citrinin, ergot alkaloids, phytoestrogens, lead, mercury. cadmium, arsenic, selenium, acrylamide, oily fish, raspberry leaf and echinacea.
1.133 Substances to be included in an overarching statement are:
- Aflatoxins, nivalenol, deoxynivalenol, T2 & HT2, patulin, vitamin E, vitamin C, camomile, peppermint, evening primrose oil, dandelion, camomile, resveratrol, heterocyclic amines, legacy pesticides, non-dioxin-like PCBs and alcohol.
1.134 Other substances that may be reviewed include dioxins, bisphenol A and fusarenon-X, some of which are awaiting the opinions of other advisory bodies. The Committee may choose to add additional substances to the list or change the approach to substances on the list as the work progresses.
Alcohol and the maternal diet: The 2016 Chief Medical Officers report
1.134 The Committee considered whether alcohol should be considered as one of the xenobiotics being considered in the review of the maternal diet. Although alcohol per se was not within the SACN remit it could be considered as a wider health issue.
As the database for the potential effects of alcohol in pregnancy was extensive, Members considered the most recent UK Government recommendations and the data on which they had been based in order to establish whether further work in this area would be of value.
1.135 The UK Government suggests that women who are pregnant or trying to become pregnant should avoid alcohol altogether. This advice, which is given on, for example, the NHS website, is based on recommendations from the “Low Risk Drinking Guidelines produced by the UK Chief Medical Officers (CMO) in 2016”. These recommendations were based on the findings of a number of systematic reviews and meta-analyses. The results of these studies were largely inconclusive with respect to the effects of low levels of alcohol exposure and methodological flaws in the studies were noted. A number of additional systematic reviews and meta-analyses have been published covering the same end points considered in the CMO report, but as previously, the results for low levels of exposure were inconclusive and methodological failings were noted.
1.136 The COM reviewed alcohol in 2005 and concluded that there was no clear evidence for a risk from (low) alcohol consumption during pregnancy, but they were not able to fully exclude a risk. The COM further concluded that alcohol itself was probably not genotoxic, however the breakdown product acetaldehyde most likely was. Overall, the COM was unclear what other chemicals may be present in alcoholic beverages that might cause an effect.
1.137 Alcohol is produced endogenously, and metabolic enzymes have been proven to be extremely effective at preventing cellular damage in the body and aiding the elimination of alcohol. Hence, the biological mechanism would need to be taken into account when considering the available epidemiology and it is possible there is a threshold for the effects of alcohol.
1.138 The CMO report is thorough and the approach and conclusions on alcohol in pregnancy are reasonable, given the data considered in the report. The evidence is not strong enough to completely rule out some risk from low levels of alcohol exposure in pregnancy. As the data published since 2011 do not greatly add to the CMO report on the clarity of the issue and given the work and resources involved, a further review would be unlikely to change the current advice to women. Members therefore agreed not to take this review further.
Ongoing topics in maternal diet
Vitamin D
1.139 The Committee assessed if exposure to excess intake of vitamin D from various sources (including UV radiation, dietary sources, and supplements) would pose a risk to maternal health.
1.140 The relationship between oral vitamin D intake and serum levels is unclear due to many uncertainties such as season, time of day, amount of skin exposed, skin pigmentation and use of SPF sunscreen. However, exposure from UV radiation is considered unlikely to result in vitamin D toxicity due to inbuilt mechanisms in the skin where pre-vitamin D3 reaches a maximum concentration in the skin within a few hours after UVB radiation exposure. Other uncertainties in the assessment is the use of data from non-pregnant women of child-bearing age (i.e., 16-49 years) to construct the exposure assessment in pregnant women, since the diet of the latter may vary.
1.141 Higher strength vitamin D supplements are likely to be the biggest contributor to vitamin D exposure, and consumption of these supplements alone is sufficient to result in exceedance of the TUL of 100 µg/day. The diet alone without consumption of vitamin D containing supplements is unlikely to be a cause of concern, and consumption of both dietary sources of vitamin D and higher strength vitamin D supplements are likely to result in exposure levels greater than the TUL of 100 µg/day.
1.142 A statement setting out the Committee’s assessment of vitamin D will be published in 2022.
Cadmium
1.143 The COT discussed a review of the literature on cadmium in the maternal diet and requested additional information, with particular regard to maternal dietary intake and the implications for subpopulations where consumption of certain food groups might be higher.
1.144 As smoking is a significant source of cadmium, information on cigarette smoke and vaping should be included also considering bystander/passive smokers. Further information on metallothionein and the role it plays in the body and the placenta was also requested.
1.145 The Committee will continue to work on cadmium during 2022.
Vitamin A
1.146 Vitamin A (retinol) is essential for the health of adults, children and developing foetuses, although both deficiency and excess lead to toxicity, particularly developmental malformation in the fetus.
1.147 Dietary retinol comes pre-formed from animal derived foods such as liver or liver products or is converted from carotenoids such as β-carotene which form the colouring matter in vegetables such as carrots and peppers. Retinol is absorbed with dietary fats, bound to plasma proteins and stored in the liver. Retinol is oxidised in the tissues to retinal, which is essential for vision, and then to retinoic acid, which is essential for fetal development and other functions.
1.148 In many countries, the issue for maternal health is deficiency but in richer nations adequate dietary levels are normally met. A tolerable upper limit (UL) of 3000 µg retinol/day has been set by EFSA as a level unlikely to cause developmental malformations but there is uncertainty about the actual level that may be associated with toxicity. However, pregnant women are advised not to consume foods such as liver or supplements such as cod liver oil which may cause them to exceed the UL.
1.149 Oral and topical retinoid-based products are used to treat severe acne, often in young women who may become pregnant and although the risks are disputed, their use is not recommended in pregnancy. Recent evidence suggests that an association between retinoid acne treatment and depression may be ill-founded, but uncertainties still exist.
1.150 Few if any ill effects have been ascribed to taking supplements containing β-carotene.
1.151 A statement setting out the Committee’s assessment of vitamin A will be published in 2022.
Ginger and ginger supplements
1.152 As part of the current programme of work on the maternal diet, the Committee considered the use of dietary supplements during pregnancy to identify those that might need reviewing. These are supplements that are not officially recommended but which are promoted by anecdotal evidence and unofficial sources as having various purported benefits. It was agreed that ginger should be considered in further detail.
1.153 The Committee considered the potential effects of ginger and ginger supplements during pregnancy and lactation, reviewing the available data on toxicity to the mother, effects on the development of the fetus or embryo, and possible interactions with drugs as well as data on potential exposure.
1.154 As it is commonly believed that ginger suppresses morning sickness, pregnant women may be using the supplements for this purpose. Whilst ginger consumption in the diet is not considered to be of concern due to the long history of safe use for culinary purposes, however, problems could arise from consumption of more concentrated products such as the various forms of supplements.
1.155 There are limited human data, and these are not strongly indicative of any toxicological concern but there are some indications of possible adverse effects and numerous uncertainties. Ginger did not appear to be systemically toxic but did appear to have reprotoxic effects at high doses in animal studies. Ginger is thought to affect prostaglandin function which may be relevant to this.
1.156 It is not possible to determine a point of departure to use in the risk assessment of ginger. While there is some equivocal evidence for the possible effect of ginger on reproduction, it is not possible to characterise it based on the data available. There is no clear indication that ginger is detrimental to consumers.
1.157 The potential for contamination of ginger with heavy metals and/or mycotoxins cannot be excluded.
1.158 A statement setting out the views of the Committee on will be published in 2022.
Iodine
1.159 As part of the work on the maternal diet, the COT was asked to consider the potential effects that excess iodine intake may have during preconception, pregnancy and lactation.
1.160 Iodine is an essential component of thyroid hormones which are important in growth and development. It is found in foods such as fish and seafoods as well as fortified products and food supplements. Seaweed is a very rich source of iodine and may lead to high levels of consumption in some consumers.
1.161 Iodine was initially discussed in 2020 and the Committee considered issues such as exposure, biomarkers and individual susceptibility to the effects of excess iodine.
1.162 Overall, members agreed that while there were no concerns in the general population, exposure to excess iodine in high seaweed consumers could pose a potential risk to maternal health. It was concluded that the currently available data was not sufficient to enable a risk benefit assessment to be performed.
1.163 The final statement setting out the Committee’s views on iodine will be published in 2022.
NAMs Roadmap
1.164 Advances in biology, computer science and other related fields are paving the way for major improvements in how environmental and public health risks posed by potentially toxic chemicals can be evaluated. The combined advances in discovery and clinical sciences, data science and technology have resulted in toxicity testing which has reached a pivotal transformation point known as the 4th industrial revolution (4IR). One of the major recent scientific advancements is the development of New Approach Methodologies (NAMs) including high throughput screening, omics and in silico computer modelling strategies such as Artificial Intelligence and machine learning for the evaluation of hazard and exposure. This also supports the Replacement, Reduction and Refinement (3Rs) approach.
1.165 The future of the safety assessment of chemicals in food depends on adaptability and flexibility in utilising the best scientific methodologies and strategies available to respond to the accelerating developments in science and technology.
1.166 NAMs are gaining traction as a systematic approach to support informed decision making in chemical risk assessment. Integration of these technologies as part of the FSA chemical risk assessment process will be fundamental in the future in the future of human safety assessments to protect consumers.
1.167 The Food Standards Agency (FSA) responds to food incidents and it is important that robust risk assessments on the safety of a chemical can be provided.
However, sometimes there is very little, or no, toxicological information for a given chemical. For such chemicals, the use of NAMs could provide a more indicative level of risk and therefore greater confidence can be provided for them as well as less uncertainty that individual compounds can be assessed.
1.168 In order to achieve this, the FSA and COT are developing a UK roadmap towards acceptance and integration of NAMs, including predictive toxicology methods using computer modelling, into safety and risk assessments for regulatory decision making.
1.169 During 2020, Members discussed the latest draft version of the roadmap The Committee endorse the roadmap and a supporting workshop, and congratulate the FSA for taking the lead in this area. Developing the roadmap will involve engaging with other government departments and regulatory bodies.
The potential health risks of bamboo bio-composite food contact materials
1.170 Advice on biobased food contact materials (BBFCMs) has been increasingly requested from the Food Standards Agency (FSA) so it was therefore considered timely for the Committee to review the available toxicological information on BBFCMs.
1.171 It was agreed that a health risk assessment should be conducted for bamboo composites based on its potential hazards.
1.172 The migration of formaldehyde and melamine from bamboo composite cups is a potential concern to human health and it would therefore be appropriate to conduct a full risk assessment once UK data are available.
1.173 As obtaining the data and providing a full risk assessment will require time, the COT agreed to publish an interim position paper to set out their concerns and allow for risk management action.
1.174 The interim position paper will be finalised and published in 2022.
Chitin and chitosan in food packaging materials
1.175 The FSA is currently assessing whether there are any risks to health posed by bio-based food contact materials (BBFCMs). One of the first materials to be reviewed was including that containing chitin or chitosan.
1.176 Chitin and chitosan can be derived from fungi or from shellfish. Therefore there are potential concerns for individuals who are allergic to shellfish, but only limited data are available.
1.177 The risk of allergenicity from these BBFCMs appears to be low. However, before the potential risks to human health can be fully assessed, it would be useful to have an indication or estimation of total exposures to allergenic proteins from BBFCMs, for example the upper bound levels of ingestion, or range of amounts of BBFCMs in contact with different foods.
1.178 Further information on chitin or chitosan derived from fungi is also needed.
1.179 Due to a scarcity of relevant data in the scientific literature, it is not currently possible to undertake a reliable exposure assessment due to the uncertainties involved.
1.180 A second draft statement on this work will be presented to the COT in 2022.
PBPK Workshop
1.181 The FSA and the COT held a “PBPK for Regulators” workshop in December 2020 in a multidisciplinary setting with delegates from regulatory agencies, government bodies, academia and industry. The workshop provided a platform to enable expert discussions on the application of PBPK to human health risk assessment in a regulatory context.
1.182 The presentations covered current applications of PBPK modelling: in the agrochemical industry for in vitro to in vivo extrapolation (IVIVE); pharmaceutical industry for drug absorption related issues (e.g. the effect of food on drug absorption) and drug-drug interaction studies, as well as dose extrapolations to special populations (e.g. those with a specific disease state, paediatric/geriatric age groups, and different ethnicities); environmental chemical risk assessment fields; an overview of the current regulatory guidance; and a PBPK model demonstration. This enabled attendees to consider the wide potential and fit for purpose of application of PBPK modelling in these fields. Attendees further considered the applicability of PBPK models in the context of future food safety assessment, for refining exposure assessments of chemicals with narrow margins of exposure and/or to fill data gaps from more traditional approaches (i.e., data from animal testing).
1.183 The overall conclusions from the workshop proceedings were as follows:
- PBPK modelling tools are applicable in the explored areas of use, and there is some expertise available for their utilisation.
- PBPK modelling offers opportunities from which to address questions for compounds that are otherwise not solvable.
- Widespread acceptance amongst regulatory bodies appears to be limited by lack of available in-house expertise.
- Familiarisation using real world case studies would help in developing more experts in the field and increasing acceptance.
- In a regulatory context, establishing fitness for purpose for the use of PBPK models requires multi-partite discussion and harmonised guidance.
- PBPK modelling is part of the wider “new approach methodologies” for risk assessment.
1.143 A summary of proceedings from this workshop will be published in due course.
Risk assessment of potential constituents and contaminants in cow’s milk
1.144 Plant-based drinks have become increasingly popular in the United Kingdom (UK) both for individuals with an allergy to cows’ milk or lactose intolerance and those who wish to avoid dairy products for other ethical or cultural reasons (see paragraph 1.30).
1.145 As noted elsewhere, following on from the assessment of plant based drinks a joint SACN/COT Working Group has been established to bring together the nutritional and toxicological aspects of plant based drinks.
1.146 The main comparator for plant-based drinks should be cow’s milk and to enable comparison, the potential chemical constituents and contaminants within cows’ milk should be reviewed. These included veterinary medicine residues, pesticide residues, nitrate and nitrite, bisphenol A, phthalates, dioxins and dioxin-like biphenyls, non-dioxin-like polychlorinated biphenyls, polycyclic aromatic hydrocarbons, selected isoflavones, heavy metals, iodine, chlorate and perchlorate, mycotoxins, naturally occurring oestrogens in cows’ milk, insulin like growth factor, per- and polyfluoroalkyl substances, brominated flame retardants and microplastics.
1.147 The Committee concluded that there were no health concerns arising from the consumption of cow’s milk associated with the compounds noted above.
1.148 A statement covering the Committee’s views on the safety of milk will be published in due course.
Interim Position paper on Titanium Dioxide
1.149 Following the discussions by both COT and COM a draft interim position paper on titanium dioxide, capturing the outcomes of the discussions from the two Committees and outlining the next steps was prepared. The Interim Position Paper will be published in due course.
COT Working Groups 2021
In this guide
In this guideSETE
Report and Guidance of the Synthesis and Integration of Epidemiological and Toxicological Evidence Subgroup (SETE) of the Committee on Toxicity and the Committee on Carcinogenicity
1.150 The UK Committees on Toxicity (COT) and on Carcinogenicity (COC) regularly review epidemiological and toxicological evidence in their risk assessments. There is, therefore, a need for guidance on the approaches used by the Committees to integrate these evidence streams, both for scientific consistency and to ensure public transparency. To that end, the Committees established the Synthesising and Integration of Epidemiological and Toxicological Evidence Subgroup (SETE) to review and document current practice and provide applicable guidance.
1.151 SETE recognised that issues on which advice from the Committees is sought varies considerably and hence the guidance proposed should be sufficiently flexible to address this.
1.152 Scoping and problem formulation were identified as the crucial first step in the process. This ensures the right questions are asked, helps make the most efficient use of resources and identifies the most appropriate approaches to use in the assessment. An established system or guidance to assess the separate/different evidence streams should be followed where feasible.
1.153 For both epidemiological and toxicological evidence, a prescriptive checklist or scoring approach is not recommended. However, identifying the strengths and weaknesses of studies is important. The decision-making process should be robust, transparent, evidence-based, defensible and documented but equally importantly, it should be easy to use. Collaboration and ongoing dialogue between epidemiologists, exposure experts and toxicologists are strongly advised. Information on mode of action (MOA) can be invaluable for evidence integration as it underpins weight of evidence considerations by providing the mechanistic link between empirical observation and biological plausibility.
1.154 All lines of evidence should be considered, with no specific hierarchy a priori. One way to clearly depict the influence of the different lines of evidence on causality is via visual representation. Decisions on whether there is sufficient information to reach a conclusion or whether a causal relationship in humans is more likely or unlikely can be reached based on where the causal interference appears on a graph. It is important to begin with the initial estimate of causal interference at the centre of the graph. Depending on whether the toxicological, mechanistic or epidemiological evidence assessed supports or discounts (or has no clear influence on) a conclusion of causality, placement on the graph is then moved accordingly, either in a positive or negative direction. The movement is influenced by several factors, including the strength or weakness of the evidence, any relative weighing given to epidemiological and toxicological studies and the uncertainties associated with the data. As more information is included in the process and/or becomes available, the placement of the toxicological and/or epidemiological evidence can be easily adjusted and the impact on any possible conclusion easily seen.
1.155 In contrast to other approaches, the above visualisation aims to provide a pictorial representation of the consensus views of a Committee on the influence of the different lines of evidence on causation, assessed by debate and agreement of scientific experts. In this way, it provides a more objective means of collating the views of the Committee and communicating the agreed conclusion of a Committee on the likelihood of causation.
1.156 The conclusion should be stated, with an estimate of the overall uncertainty and, where appropriate, guidance on how data gaps could be filled.
1.157 The full SETE report and guidance document (Annex 1) can be found on the COT website: SETE Outputs.
Please note, the guidance will be trialled by the COT for 2 years and then reviewed.
Plant based drinks
1.158 Plant-based drinks have become increasingly popular in the United Kingdom (UK) both for individuals with an allergy to cows’ milk or lactose intolerance and those who wish to avoid dairy products for other ethical or cultural reasons. Three such drinks have been reviewed by the Committee – see paragraph 1.130.
1.159 SACN have also considered these drinks from a nutritional perspective. To bring these two strand together, a joint Working Group had been established. The Working Group started work In December 2021 and it is hoped that it will report in 2022.
2021 Membership of the Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment
In this guide
In this guideChairman:
Professor Alan Boobis OBE, PhD, FBTS, FBPhS
Emeritus Professor of Toxicology in the Faculty of Medicine at Imperial College London.
Members:
Dr Phil Botham BSc, PhD
Principal Science Advisor at Syngenta (part time).
Ms Jane Case
Lay Member.
Dr Stella Cochrane BSc PhD
Science Leader for Allergy and Immunology in Unilever’s Safety and Environmental Assurance Centre.
Dr James Coulson BSc MBBCh Dip Med Tox Dip Therapeutics LLM MD FRCP FRCPE ERT
Clinical Reader at Cardiff University, Honorary Professor in Clinical Pharmacology and Toxicology, Cardiff Metropolitan University, Honorary Consultant Physician, Clinical Pharmacologist and Toxicologist, Cardiff & Vale University Health Board.
Dr René Crevel
Director, René Crevel Consulting Limited.
Dr Caroline Harris PhD, CChem, FRSC
Practice Director and Principal Scientist, Exponent International Ltd.
Professor Gary Hutchison
Dean of Applied Sciences at Edinburgh Napier University, with responsibility for Life Sciences, Social Sciences, Psychology, Teacher Education and Sports Exercise and Health Sciences.
Dr Sarah Judge BSc, PhD
Lecturer in Pharmacology in the School of Biomedical, Nutritional and Sport Sciences at Newcastle University.
Dr Gunter Kuhnle
Professor of Nutrition and Food Science.
Dr David Lovell
Emeritus Reader in Medical Statistics at St George’s Medical School, University of London.
Dr Mac Provan
Director of Regulatory Science Ltd.
Ms Juliet Rix
Lay Member.
Dr Michael Routledge
Associate Professor of Environmental Toxicology in the School of Medicine at Leeds.
Dr Cheryl Scudamore
RCVS Specialist in Veterinary Pathology (laboratory animals) working as independent consultant in experimental and toxicological pathology.
Dr Natalie Thatcher
Mondelēz International.
Professor Mireille Toledano
Chair in Perinatal and Paediatric Environmental Epidemiology, Faculty of Medicine, School of Public Health, Imperial College London.
Professor Faith M Williams MA PhD hon FBTS (until March 2021).
Emeritus Professor of Toxicology, Medical Toxicology Centre and Institute of Cellular Medicine, Newcastle University.
Professor Philippe Wilson
Professor of Animal Science and Bioinformatics, Nottingham Trent University, and Head of Conservation at the Rare Breeds Survival Trust.
Professor Matthew Wright BSc, PhD
Professor of Toxicology, Institute of Cellular Medicine, Newcastle University.
Professor Maged Younes
Independent expert on toxicology and biochemical pharmacology.
Professor Thorhallur I. Halldorsson (from April 2021).
Professor at the Faculty of Food Science and Nutrition at the University of Iceland.
Dr Simon Wilkinson (from April 2021).
Senior Lecturer in Pharmacology in the School of Biomedical, Nutritional and Sports Sciences at Newcastle University.
Professor Shirley Price (from April 2021).
Emerita Professor of Toxicology at the University of Surrey.
Secretariat
Ms Catherine Mulholland BSc (Hons), ERT Scientific Secretary
Ms Britta Gadeberg BSc (Hons) MSc Scientific Secretary – PHE
Dr David Gott BSc (Hons) PhD
Dr Alexander Cooper BSc (Hons) MSc PhD
Dr Barbara Doerr BSc (Hons) MSc PhD
Dr Douglas Hedley BSc (Hons) MSc PhD
Ms Frances Hill BSc (Hons) MSc (until May 2021)
Ms Jocelyn Frimpong Manso BSc (Hons) MSc
Ms Cleanncy Hoppie BSc (Hons) MSc
Mr Barry Maycock BSc (Hons) MSc
Dr Olivia Osborne BSc (Hons) (Exon) PhD
Ms Claire Potter BSc (Hons) MSc ERT
Dr Joseph Shavila BSc (Hons) MSc PhD
Ms Chloe Thomas BSc (Hons) (until April 2021)
Ms Sabrina Thomas BSc (Hons) MSc
Ms Chara Tsoulli BSc (Hons) MSc Ms
Frederique Uy BSc (Hons) MSc
Miss Sophy Wells
Dr Gaetana Spedalieri (from September 2021)
Mr Thomas Hornsby BSc (Hons) MSc (from July 2021)
Mr Lawrence Finn BSc (Hons) MSc (from October 2021)
Ms Gail Drummond BSc (Hons) MSc, LLB, PG Dip (law) (from June 2021)
Dr Emily Hudson BSc (Hons) Mres (from November 2021)
Dr David Kovacic (from October 2021)
Declaration of COT members interests during the period of this report 2021
In this guide
In this guideProfesso r Alan Boobis OBE, PhD, FBTS, FBPhS
|
Personal Interest |
Employee: Imperial College London, Department of Medicine (retired June 2017, part-time appointment from Aug 2017-May 2019). Full retiral June 2019. Emeritus Professor of Imperial College London, National Heart & Lung Institute. |
|
Personal Interest
|
Membership: ILSI & ILSI,HESI Board of Trustees ILSI Europe. Board of Directors Science Advisory Board of Swiss Centre for Applied Human Toxicology. Dept. of Health Committee on the Medical Effects of Air Pollutants WHO/FAO JMPR. WHO/FAO JECFA (vet). WHO TobReg. WG10 TC126 (Intense Machine- smoking Regime for Testing Cigarettes). EUROTOX. British Pharmacological Society, British Toxicology Society, Society of Toxicology (USA). Royal Society of Biology (until 2017). Michigan State University MSU Center for Research on Ingredient Safety (CRIS) (External Advisory Committee). Agency for Innovations in Food and Chemical Safety Programme. Science, Technology and Research, Singapore (A*STAR) (Scientific Advisory Board). |
|
Non Personal Interest |
None. |
Dr Phil Botham
|
Personal Interest |
Employee: Syngenta - Principal Science Advisor (part time). |
|
Personal Interest |
Shareholder: AstraZeneca, Regulatory Science Associates (Part, Time Consultant). |
|
Personal Interest |
Membership: British Toxicology Society, Society of Toxicology (USA), European Centre for Ecotoxicology and Toxicology of Chemicals Scientific Committee, European Crop Protection Association Toxicology Expert Group, Crop Life International Human Health Steering Team. |
|
Non-Personal Interest |
None. |
Ms Jane Case
|
Personal Interest |
Employee: Company Secretary of Muse Interiors Stevens & Bolton LLP (as Jane Hughes). |
|
Personal Interest |
Membership: None. |
|
Personal Interest |
Shareholder: Standard Life Santander |
|
Non-Personal Interest |
None. |
Dr Stella Cochrane
|
Personal Interest |
Employee: Unilever. |
|
Personal Interest |
Membership / Affiliation: Unilever representative on the UK FDF Allergen Steering Group (Deputy Chair), FDE Allergen Group and University of Nebraska Food Allergy Research & Resources Board. |
|
Personal Interest |
Shareholder: Unilever. |
|
Non-Personal Interest |
None. |
Dr James Coulson
|
Personal Interest |
Employee: Cardiff University, Director of Medical, Scientific and Toxicology Consultancy Ltd. |
|
Personal Interest |
Membership: British Medical Association, British Pharmacology Society, British Toxicology Society National Trust, Royal College of Physicians of London. |
|
Non-Personal Interest |
None. |
Dr René Crevel
|
Personal Interest |
Consultant: Réne Crevel consulting. |
|
Personal Interest |
Membership/affiliation: ILSI Food Allergy Task Force: Chair. |
|
Personal Interest |
Shareholder : Unilever, Centrica, BG Group, National Grid, Lloyds. |
|
Non-Personal Interest |
None. |
Professor Thorhallur Ingi Halldorsson COT Member from June 2021.
|
Personal Interest |
Employee: Faculty of Food Science and Nutrition, University of Iceland. |
|
Personal Interest |
Membership: European Food Safety Authority - Scientific committee and various working groups. Nordic Council of Ministers - revision of the 2022 Nordic Nutrition Recommendation). Icelandic Risk Assessment Committee for Food, Feed, Fertilizers and Seeds (IRAC) – occasional expert work. The Nutricia Research Foundation – review of applications once a year. The Icelandic Research Found (RANNIS) – occasional member of different expert panels. |
|
Non-Personal Interest |
None. |
Dr Caroline Harris
|
Personal Interest |
Employee: Exponent International Ltd. |
|
Personal Interest |
Membership: International Union of Pure and Applied Chemistry. |
|
Personal Interest |
Shareholder: Exponent Inc. |
|
Personal Interest |
Fellowships: Royal Society of Chemistry. |
|
Non-Personal Interest |
Membership: Expert Committee on Pesticides. |
Professor Gary Hutchison
|
Personal Interest |
Employee: Dean of Applied Sciences at Edinburgh Napier University. |
|
Personal Interest |
Membership: Hazardous Substances Advisory Committee DEFRA, British Toxicology Society. |
|
Non-Personal Interest |
None. |
Dr Sarah Judge
|
Personal Interest |
Employee: Newcastle University, Lowcock Properties Ltd. |
|
Personal Interest |
Membership: British Pharmacology Society, British Toxicology Society International Association for Neurotoxicology. |
|
Non-Personal Interest |
Research Funding. |
Professor Gunter Kuhnle
|
Personal Interest |
Employee: Professor of Nutrition and Food Science, University of Reading. |
|
Non-Personal Interest |
None. |
Dr David Lovell
|
Personal Interest |
Employee: Reader in Medical Statistics, St Georges Medical School University of London. |
|
Personal Interest |
Membership: HESI GTTC – Biometrics Society, British Toxicology Society Genetics Society, Royal Society of Biology Laboratory Animal Science Association, Royal Statistical Society Statisticians in the Pharmaceutical Industry, United Kingdom Environment Mutagen Society (UKEMS), UK National Centre of Replacement, Refinement and Reduction of Animals in Research (NC3Rs), MRC EMINENT Scientific Review Board.
Also, private member of: British Trust of Ornithologists (BTO) English Heritage, Liberty, Campaign of the Protection of Rural England (CPRE), Kew Gardens, Sandwich Bay Bird Observatory Trust (SBBOT), Chelsea Physic Garden, National Trust. |
|
Personal Interest |
Shareholder: National Grid, Pfizer, AstraZeneca (spouse shareholder), National Grid plc (spouse shareholder). |
|
Non-Personal Interest |
None. |
Professor Shirley Price COT Member since June 2021.
|
Personal Interest |
Employee: None. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
Trusteeships: Gas Safety Trust |
|
Non-Personal Interest |
Other: I can confirm that as the President of the British Toxicology Society (BTS) I hold a non-personal and non-specific interest in both GSK and AstraZeneca on the Society’s behalf. These non-personal and non-specific interests relate to donations provided by both companies to the British Toxicology Society (BTS) to support their Annual Congress and Education and Training. |
Dr Mac Provan
|
Personal Interest |
Employee: Director of Regulatory Science Ltd. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Ms Juliet Rix
|
Personal Interest |
Employee: None. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Dr Cheryl Scudamore
|
Personal Interest |
Employee: Independent consultant in experimental and toxicological pathology. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Dr Natalie Thatcher
|
Personal Interest |
Employee: Mondelēz International. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Professor Mireille Toledano
|
Personal Interest |
Employee: Marit Mohn Chair in Perinatal & Paediatric Environmental Epidemiology, Imperial College London. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Dr Simon Wilkinson COT Member since June 2021.
|
Personal Interest |
Consultancies and other fee-paid work: Consultancy for L’Oreal, Paris. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Professor Philippe Wilson
|
Personal Interest |
Employee: Nottingham Trent University, Rare Breeds Survival Trust. |
|
Personal Interest |
Membership: None. |
|
Non-Personal Interest |
None. |
Professor Matthew Wright
|
Personal Interest |
Consultancies and Direct Employment: Newcastle University. |
|
Personal Interest |
Membership: British Toxicology Society, Society of Toxicology (US), EFSA FAF Panel. |
|
Personal Interest |
Miscellaneous: Toxicology – Associate Editor. |
|
Non-Personal Interest |
Support by Industry: GSK, Lubrizol. |
Professor Maged Younes
|
Personal Interest |
Employee: Independent expert in toxicology and biochemical pharmacology. |
|
Personal Interest |
Membership: Chair of EFSA ANS panel, Chair Commission on evidence-based methods in risk assessment, Federal Institute for Risk Assessment (BFr), Germany. Society of Toxicology, USA German Society of Experimental and Clinical Pharmacology and Toxicology. Society for Risk Analysis. |
|
Non-Personal Interest |
None. |
Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment- Preface 2021
In this guide
In this guide
Head and shoulders image of Professor Gareth Jenkins in front of a light-coloured background wearing a dark coloured shirt.
I am pleased to present this report on the work of the Committee on Mutagenicity (COM) during 2021. I was honoured to be asked to take over the role of Chair of COM in May 2021 and I would like to begin by paying tribute to my predecessor (Dr David Lovell) for his stewardship of COM during the preceding years and Chairing the February 2021 meeting.
The Committee on Mutagenicity (COM) provides advice on potential mutagenic activity of specific chemicals at the request of UK Government Departments and Agencies. Such requests generally relate to chemicals for which there are incomplete, non-standard or controversial data sets for which independent authoritative advice on potential mutagenic hazards and risks is required. Recommendations for further studies are, on occasions, made.
The Committee also advises on important general principles and on new scientific work related to the assessment of mutagenic risk and makes recommendations on wider aspects of mutagenicity testing. The membership of the Committee, declarations of their interests, agendas and minutes of meetings, and statements are all published on the internet. Latest from the Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment
In 2021, the updated COM guidance on genotoxicity testing strategy was published (MUT/2021/01). This update, begun in 2020, sets out the suggested strategy for genotoxicity testing of chemicals and updates our position to consider advances in the field of safety testing. COM also updated guidance on testing of germ cell mutagens (MUT/2021/02) and the use of 3D tissue models as alternative approaches to animals in testing (MUT/2021/03). The documents will be published on the COM website. The 3D tissue strategy responds to the growing focus on animal alternatives driven by the production of novel sophisticated tissue models which can recapitulate aspects of human biology.
In 2021, COM discussed the safety testing of impurities (MUT/2021/04) and the use of QSAR and toxicogenomics in testing (MUT/2021/05 and MUT/2021/06).
In 2021, COM started a discussion of the genotoxicity of titanium dioxide (MUT/2021/07 and MUT/2021/12), following the updated opinion published by EFSA in 2021. This review of titanium dioxide will be continued in 2022.
In 2021, COM further discussed the use of toxicogenomics in safety testing (MUT/2021/08), separating out the transcriptomics aspect from the next generation sequencing (NGS) approaches. Given the advances in NGS in general, it is likely that over the coming years, NGS approaches may replace some traditional mutation testing platforms. COM also published guidance on a testing approach for nanomaterials, with a focus on considerations of the fact that key physico-chemical aspects of nanomaterials render some traditional genotoxicity tests not suitable (MUT/2021/09).
COM also discussed the potential genotoxicity of specific compounds as requested by Government departments and agencies. For example, COM reviewed the genotoxicity of Hydroxyanthracene Derivatives (MUT/2021/12) and associated human health risks.
The Committee carried out its annual Horizon scanning exercise, identifying potential topics for future work. The COM continues to be interested in hearing from Government Departments and Agencies on how its advice is acted upon.
The COM maintained its awareness of the implications of EU EXIT on its work and remained alert to the continuing uncertainty as to how the UK's regulatory environment and its relationships with international organisations will develop in 2022 and onwards.
I would specifically like to thank the COM secretariat for their exceptional support to the COM and to the WRc/IEH team for the excellent work they delivered in 2021. As always, I am grateful for the support of the individual members of the committee for their expert advice, the effort and time they put in and their support throughout the year.
Professor Gareth Jenkins
COM Ongoing Work 2021
In this guide
In this guideCOM Guidance Series Update
2.1 The updating of the overarching COM Guidance document on a strategy for the genotoxicity testing of chemicals was finalised in 2021. Amendments to the overarching COM Guidance document had previously been considered at Committee meetings in July 2018 (paper MUT/2018/09), October 2018 (paper MUT/2018/13), February 2019 (MUT/2019/01), October 2019 (MUT/2019/12), February 2020 (MUT/2020/03), June 2020 (MUT/2020/09) and November 2020 (MUT/2020/16). An additional sub-group meeting was held in January 2021 to complete review of comments left outstanding following the November 2020 meeting.
2.2 Following consideration of paper MUT/2021/01 the update of the overarching COM Guidance document on a strategy for the genotoxicity testing of chemicals was agreed by members, signed off by Chair action and published on the COM website. It was intended that this would be updated in the future as part of a rolling revision.
Guidance Statement – Germ Cell Mutagens
2.3 Drafts of a stand-alone guidance statement on genotoxicity testing strategies for germ cell mutagens were considered at the Committee meeting in February 2019 (MUT/2019/05), in October 2019 (MUT/2019/12), in June 2020 (MUT/2020/11) and November (MUT/2020/17). In 2021, members considered paper MUT/2021/02, which presented changes suggested following the November 2020 meeting. Following agreed amendments, the finalised document was signed off by Chair’s action and published on the COM website.
Guidance Statement – 3D Models
2.4 Drafts of a stand-alone guidance statement on the use of 3D models for genotoxicity testing were considered at the Committee meetings in February 2019 (MUT/2019/04), October 2019 (MUT/2019/12), June 2020 (MUT/2020/11) and November (MUT/2020/18). In 2021, members considered paper MUT/2021/03, which included suggested changes following the meeting in November 2020. Following agreed amendments, the finalised document was signed off by Chair’s action and published on the COM website.
Guidance on Genotoxicity Testing Strategies for Nanomaterials
2.5 Genotoxicity testing of nanomaterials (NMs) was recognised by the Committee as a rapidly developing area. Paper MUT/2021/09 presented a draft COM Guidance on the genotoxicity testing strategy for NMs. This was prepared to a format previously agreed by COM at the meeting in November 2020 (MUT/2020/19). Members considered that it was important to add a note to clarify that ‘Stage 0’ of the COM recommended approach for genotoxicity testing would not apply to NMs. A question was raised regarding whether COM should recommend a positive control for NM testing. This was not considered feasible at present as this would probably need to be both assay and cell line specific, due to differing sensitivities. Members requested that this information be added to the document. It was also agreed that a note should be added to consider the most appropriate dispersion technique for a specific NM. Following these amendments, members agreed that a final version of the document could be signed off by Chair’s Action and published on the COM website. It is recognised by the Committee that this is a rapidly developing area and updates will be carried out as new information becomes available.
Guidance Statement on Testing for Impurities – Update
2.6 The COM published a guidance statement in 2012 on a strategy for genotoxicity testing and mutagenic hazard assessment of impurities in chemical substances. Since 2012, there have been a number of initiatives in this area and as part of the ongoing update of the COM Guidance Statement series, members agreed that the Guidance document should be updated. A draft revised document was presented at the Committee meeting in November 2020 (MUT/2020/21) and following comments and suggestions from members a revised draft statement was produced (MUT/2021/04) and presented at the February 2021 meeting. During review it was suggested that the impurities guidance statement and QSAR guidance statement could be merged as there was overlap between the two areas.
COM Guidance Statement on the Use of QSAR Models
2.7 A range of Quantitative Structure-Activity Relationship (QSAR) models have been developed to predict genotoxicity. The COM has previously agreed that where no genotoxicity data are available, the intrinsic chemical and toxicological properties of a chemical must be considered prior to developing a genotoxicity testing programme, as reported in “Guidance On A Strategy For Genotoxicity Testing Of Chemical Substances” (COM, 2011) and as updated in 2021. This guidance describes a staged approach to testing consisting of stages 0 (preliminary considerations including physico-chemical properties), 1 (in vitro genotoxicity tests) and 2 (in vivo genotoxicity tests). QSARs are incorporated into Stage 0 of the COM guidance.
Alternatives to animal testing and the usefulness of computational methods in the prediction of genotoxicity are areas of increasing research. QSAR models and their predictions currently cannot replace the need to undertake the in vitro and in vivo genotoxicity tests required to derive conclusions on mutagenic hazard except in specific regulatory settings. As the development and use of QSAR is a rapidly developing field, it was agreed that the current text in the COM overarching guidance document should be reduced and a larger ‘stand-alone’ guidance statement be prepared which could be updated as needed.
2.8 A draft document - ‘Guidance Statement on the use of QSAR models to predict genotoxicity’ was prepared and discussed by COM in February 2019 (MUT/2019/03). Following amendments, a revised paper was discussed in February 2020 (MUT/2020/02) and November 2020 (MUT/2020/20). No agreement was reached as to whether the draft guidance statement was ‘fit-for-purpose’, and it was also suggested that QSARs could be incorporated into the COM guidance on impurities, as this is where it is likely to be used.
2.9 Following a further draft COM Guidance on QSARs (MUT/2021/05) considered at the February 2021 meeting, a sub-group discussion with some COM members was held in September 2021 to plan a way forward. It was suggested that, based on current acceptance and use of QSARs, incorporation of examples of use and reporting of data should be included in the updated impurities guidance document, with a link to the OECD portal provided to give the most current perspective/tools etc. A more general description (taken from the current draft document) would then be re-introduced into the COM overarching guidance document to support the Stage 0 testing text.
2.10 Members agreed that it was important for any COM guidance to highlight applications of QSAR, rather than providing a list of QSAR models and approaches.
Toxicogenomics and Risk Assessment: Application of Transcriptomics and Next Generation Sequencing to Genotoxicity and Carcinogenicity Assessment
2.11 At the COM meeting in February 2021, during discussions of some preliminary literature on ‘toxicogenomics and risk assessment’ (MUT/2021/06), members noted that this field could at present be considered to comprise two different major elements; the more highly established field of transcriptomics, and the newer area of next-generation sequencing technologies. It was felt that it would be useful for a document to be prepared providing a preliminary overview of these two areas and their potential applications to risk assessment in the fields of mutagenicity and carcinogenicity. Discussion paper MUT/2021/08 provided an overview of these two areas, summarising narrative from three recently published review articles.
2.12 Members noted that overall, this was a fast-developing area. For this reason, it may be difficult for the COM to establish a specific guidance document, as this would rapidly become out of date. However, members also considered that this is a very important area in the development in genotoxicity assessment and should be kept under evaluation by the Committee.
2.13 Some major areas of work in this field were highlighted. These included: Current efforts to obtain mutational signatures and match these to environmental exposures, which was noted as an area that the COM would probably wish to focus on further; Progression of work on TGx-DDI (a transcriptomic biomarker for genotoxicity), noting that data is being passed to regulators with the aim to be able to provide guidance; Development of duplex sequencing at Health Canada, which is starting to be useful for investigations of germ-cell mutagenesis and for dose-response analysis; Use of cancer-driver mutations via the ‘CarcSeq’ method at FDA.
2.14 In terms of document progression, a more detailed paper could be envisaged, noting techniques and methodologies that are becoming available, and describing some examples of how these techniques may be becoming applicable to investigation of genotoxicity. It was agreed that further development of any paper from COM concerning the use of toxicogenomics for risk assessment purposes would be discussed by a small sub-group of interested members.
Presentation by Professor Michael K Skinner – Washington State University, USA – Environmental Toxicant Induced Epigenetic Transgenerational Inheritance of Disease. Generational Toxicology – Open to COC and COT Members
2.15 At the February 2021 meeting, Professor Skinner from Washington State University (Washington, USA) presented a talk entitled ‘Environmental Toxicant Induced Epigenetic Transgenerational Inheritance of Disease: Generational Toxicology’. This was also open to COC and COT members.
2.16 As an introduction, Professor Mike Skinner highlighted that it is difficult to explain all disease based solely on the genome and that that environmental factors also play a role on the occurrence of disease. What is observed is not completely explained by the paradigm of the genome affecting gene expression, which in turn affects physiology and the development of disease. For example, the development of disease in identical twins is reported to vary when identical twins live in different regions. This indicates that other factors are involved in addition to individual DNA sequence.
2.17 Professor Mike Skinner summarised animal studies that showed adverse effects in future generations (i.e., F2 and later generations, where the germline was not directly exposed to the initial test chemical) arising from an initial chemical exposure in pregnant females. The observed adverse effects arose from epigenetic changes. Epigenetic effects could arise from chemical induced changes in DNA methylation, histone modifications and effects on RNA (i.e., not involving a change in the DNA sequence). Such chemical induced epigenetic changes can result in modification of gene expression.
2.17 Professor Skinner noted that if a gestating F0 female animal is exposed to a particular chemical, then the F3 generation would be first generation that did not receive a direct test chemical germline exposure. Chemical induced effects seen in the F3 generation and subsequent generation could be due to epigenetic effects or inherited changes in gene expression arising from the initial gestating exposure of the F0 female. This would be an example of transgenerational inheritance. If a non-pregnant female or a male animal was exposed to the test chemical, then the F2 generation would be the first generation that did not receive direct germline chemical exposure. Chemical induced effects in this generation could arise from inherited epigenetic changes (this would be an example of transgenerational inheritance).
2.18 A number of examples of results of chemical exposure in animals were reported where 90% of treated animals showed adverse effects in the F3 generation resulting from an initial F0 gestating female exposure. For example, vinclozolin (agricultural fungicide), TCDD/Dioxin, DDT, bisphenol A and diethyl hexyl phthalate produced adverse effects in the F1 generation and in the F3 generation. Flutamide (anti-androgenic pharmaceutical) produced adverse effects in F1, but not in F3 generation. However, atrazine (agricultural herbicide) and glyphosate (herbicide) did not induce adverse effects in F1 but did in F3 (transgenerational effect). Examples of chemically induced transgenerational disease effects included spermatogenic defects, male infertility, prostate disease, premature ovarian failure, ovarian polycystic ovarian disease, birth defects, kidney disease, obesity, behavioural effects and immune effects.
2.19 Other types of exposures can also induce epigenetic and transgenerational effects, such as extreme temperature, drought, high fat diet or caloric restriction, smoking and alcohol. Studies were described where various transgenerational epimutations and clusters were detected in the sperm genome in the F3 generation following initial chemical exposure, such as with vinclozolin and DDT.
2.20 One of the most sensitive periods of exposure is during fetal gonadal sex determination when the germ line is undergoing epigenetic programming and DNA re-methylation occurs. The suggestion that environmental toxicants can re-programme the germ line to induce epigenetic transgenerational inheritance of disease, is a new paradigm in disease aetiology, and indicates the need to assess generational toxicology in the future.
2.21 Key take home messages from the presentation included: the germline (eggs and sperm) are where epigenetic changes are critical because they get passed on in a transgenerational manner; this epigenetic transgenerational inheritance does not involve an inherited change in the DNA sequence; and a recommendation that adverse transgenerational effects need to be investigated in chemical health risk assessment. It was suggested that animal studies would be required to do this because current in vitro studies would not be suitable.
2.22 In discussions following the presentation, clarification was sought by members around how assessment of intragenerational effects may be included in current testing regimes. At the present time this can only be achieved through laboratory animal studies where the third generation needs to be evaluated, with minimum study length of between 1 and 1.5 years. It is not feasible to assess the germ cells of affected individuals because the shifts in developmental programming need to be established before the effects of the exposure are seen. A large proportion of the changes seen in earlier generations are due to direct exposure.
2.23 At present, transgenerational effects have been shown for many toxic compounds and so such testing is likely to be needed on a routine basis. There are no in vitro approaches that are effective to replace in vivo assays. It was considered possible that thresholds existed for the level of DNA methylation sites, below which long-term disease was avoided.
2.24 Diet was discussed as a major factor that had previously been linked with epigenetic changes. For a generational impact to occur the dietary influences have to be quite severe (for example, calorific restriction or high fat diets), with small shifts in diet not having an impact. Timing of exposure was also found to be key, with exposure during the early fetal life period being critical. Environmental toxicants were considered to have an effect at similar levels to calorific restriction. The importance of epidemiology studies in supporting animal data and showing causality was also discussed. Epigenetic biomarkers are needed for use in epidemiological studies, and these have not been developed.
2.25 The Chair thanked the speaker on behalf of the Committee for an interesting and informative presentation. In conclusion, it was agreed that the COM would keep an active watching brief on developments in the area, particularly in relation to inclusion in toxicity testing regimes.
Presentation on Toxicogenomics in Toxicology Testing by Dr Scott Auerbach, Division of the National Toxicology Program, National Institute of Environmental Health Sciences, USA
2.26 At the June 2021 COM meeting, Dr Scott Auerbach provided a presentation on toxicogenomics in toxicology testing. Dr Auerbach noted that functional omics technologies are a powerful tool for the characterisation of chemical effects in biological systems. Historically the primary use of omics technologies, transcriptomics in particular, has been to characterise chemical mode of action to understand toxicological mechanisms and human relevance. More recently effort has been put into use of transcriptomics as a means to identify a biological effect point of departure that roughly approximates a point of departure derived from much more resource intensive studies such as the two-year cancer bioassay.
2.27 The presentation discussed how transcriptomics has been used for qualitative characterisation of chemical effects and how it is being modelled to derive a genomic-based point of departure. In addition, some of the current scientific challenges that need to be addressed to facilitate more widespread use of genomic point of departure values for health-based guidance value determination were also discussed.
2.28 Following the presentation, the sensitivity of the methodology was queried as some genotoxic compounds may not have a strong genotoxicity signal over the shorter exposure time. This is addressed by the inclusion of doses of test substance up to the maximum tolerated dose during screening which should produce a signal if it is genotoxic. The limitation of precision of toxicogenomics in its ability to determine what proportion of cells are affected to produce the measured ‘fold’ change was highlighted. This was anticipated to be a chemical specific issue as those only affecting a small number of focal points (e.g., nitrosamines) would take longer to produce a signal than chemicals affecting multiple sites (e.g., 3,3',4,4'-Tetrachloroazobenzene) and should be taken into account to avoid inaccuracies. The use of gene-set dose response data (as a point of departure) with benchmark dose modelling was also discussed. There is no standard model to use with such data as the adverse effect size (BMR) for a particular gene is not known for many chemicals. It is also not possible at this time to take into account the effect of co-variables, which is an important consideration for human data, however this is being actively addressed by a number of groups.
Presentation on OECD development of the Mini-Ames
Dr Robert Smith, Covance
2.29 Dr Robert Smith, the UK representative on the OECD expert group developing the mini-Ames test, gave a presentation and summary of the activities of the OECD expert group on the miniaturised bacterial mutation assay.
2.30 New approaches to the or Ames test (OECD TG 471) are being explored, such as miniaturised assays, as they offer higher throughput with a significant reduction in the amount of test material required, resources and cost.
2.31 Several miniaturised versions have been developed and are already extensively used for screening purposes during product development/candidate selection or for impurity assessment/qualification. These have some differences when compared to the standard Ames assay and are not described in any existing OECD Test Guideline. Differences include the use of multi-well plates, use of liquid media rather than agar plates, the number of bacterial strains used, and the use of reduced numbers of bacterial cells (and volumes, etc.).
2.32 Following the presentation, members considered the possibility that data obtained from Ames IITM assays run by inexperienced laboratories may have influenced the findings of the Detailed Review Paper (DRP). However, there had been a requirement for laboratories to show proficiency prior to submitting data for inclusion. Although there was good concordance between the 4 assays evaluated (6 and 24-well agar plates, micro-fluctuation and Ames IITM assays) there was some remaining discussion around comparison of top doses, as the microfluctuation assay expressed doses as µg/ml and the Ames assay as µg/plate. It was also considered that exposure might be enhanced for the fluctuation assay, as fewer cells are present. The effect of pre-incubation in the fluctuation assay was queried and had been associated with a small increase in sensitivity and specificity. The maximum limit on concentration per well/plate was considered by members to be a critical factor for take-up of the assays once finalised. The OECD had produced a DRP on the evaluation of various mini-Ames assays cited in the literature compared with the standard Ames test. The OECD DRP was circulated to COM members for comment.
COM Evaluations 2021
In this guide
In this guideReview of the EFSA Opinion on Titanium Dioxide (E171) Presented by the Food Standards Agency
2.33 The Food Standards Agency requested advice from the COM on the genotoxicity of Titanium Dioxide, following a re-evaluation by the European Food Safety Authority (EFSA) published in 2021.
2.34 Titanium dioxide is an authorised Food Additive in the EU and under GB Food Law (retained EU law Regulation No 1333/2008 on food additives). It is used in food as a colour to make food more visually appealing, to give colour to food that would otherwise be colourless, or to restore the original appearance of food.
2.35 Titanium dioxide has been the subject of multiple safety evaluations. Following a review of Titanium dioxide specifications in 2019 and based on the fraction of nanoparticles present in E171, it was considered that the food additive fell under the scope of the EFSA guidance on nanotechnology and a recommendation for re-assessment of the safety of Titanium dioxide was proposed.
2.36 In the most recent evaluation published in 2021, data evaluated was for the food additive Titanium dioxide E171 as well as titanium dioxide other than E171 containing a fraction of nanoparticles <100nm or nano titanium dioxide. Concerning the genotoxicity studies, combining the available lines of evidence, the EFSA Panel on Food Additives and Flavourings (FAF) concluded that Titanium dioxide particles have the potential to induce DNA strand breaks and chromosomal damage, but not gene mutations. No clear correlation was observed between the physico-chemical properties of Titanium dioxide particles – such as crystalline form, size of constituent particles, shape and agglomeration state – and the outcome of in vitro or in vivo genotoxicity assays (i.e. a cut-off value for Titanium dioxide particle size with respect to genotoxicity could not be identified). The EFSA FAF Panel concluded that several modes of action (MOA) may operate in parallel and the relative contributions of the different molecular mechanisms resulting in the genotoxicity of Titanium dioxide particles are unknown. Based on the available data, no conclusion could be drawn as to whether the genotoxicity of Titanium dioxide particles is mediated by a mode (s) of action with a threshold(s). Therefore, the EFSA FAF Panel concluded that a concern for genotoxicity of Titanium dioxide particles cannot be ruled out.
2.37 The COM were requested to consider paper MUT/2021/03, which summarised the EFSA 2021 evaluation and included a number of questions that the COM were requested to consider.
2.38 The COM had concerns over the quality and robustness of some of the studies considered by EFSA to draw its conclusions and noted that the overall data considered by EFSA was heterogenous (e.g. the range of particles evaluated was diverse; different types of approach and assays; different doses; different cell models; some studies were published in obscure or non-genotoxicity journals and the inclusion of non-GLP studies, which all contributed to the difficulty in making comparisons and an overall evaluation). Members were also concerned over the potential for publication bias in the studies evaluated by EFSA (i.e. where negative studies were less likely to be published). It was also noted that until relatively recently, the specification of E171 was poorly defined, which contributed to uncertainty and difficulty in evaluation.
2.39 Regarding mode of genotoxic action, the COM agreed that the evidence indicated an indirect interaction with DNA with a threshold for genotoxicity. Some positive results were found with a mixture of nano and micro particles. It was impossible to interpret which fraction was responsible, although pure micro sized particles generally were negative. The in vivo studies tended to be of better quality and negative. The nano-fraction in E171 is thought to be low but the fraction of nanoparticles (<100nm) can be over 50%. The percentage of the nano-fraction and its bioavailability are important factors when considering risk assessment.
2.40 Members considered that the lack of quality in the evidence (e.g. mixed particle sizes (micro and nanoparticles) and a wide variety of testing approaches) did not allow definitive conclusions to be drawn and therefore did not agree with the EFSA overall conclusions on the genotoxicity of E171 Titanium dioxide. A review of more reliable and robust dataset may be required before conclusion could be drawn on the mutagenicity of titanium dioxide particles. Members noted that EFSA made no clear distinction between the genotoxicity of nano-sized and micro-sized titanium dioxide 2.41 particles. EFSA seemed to have put a lot of emphasis on the evidence from nano-sized particle studies when nanoparticles made up only a small fraction of E171. The COM suggested that if practicable, restricting the amount of nanoparticles in the specification for E171 may reduce any potential genotoxicity risk. Additionally, the COM considered that the wording of EFSA’s conclusion was not helpful from a risk communication perspective. Due to the heterogenous data and equivocality of the evidence further refinement of the data evaluated may be needed before definitive conclusions on the genotoxicity and safety of titanium oxide could be made. Currently, the EFSA conclusions were not justifiable based on the available evidence and this may create unnecessary concern for the public.
2.42 The COM agreed to develop an approach to evaluating all the available data (e.g., sifting for relevant and suitable studies) before continuing its review of the genotoxicity of titanium dioxide and before it could derive any firm conclusions or opinion.
Hydroxyanthracene derivatives
2.43 On the request of the UK-wide Nutrition Labelling Composition and Standards (NLCS) policy group, the UK Food Standards Agency (FSA) commissioned an independent view from the COM to advise on the genotoxicity of hydroxyanthracene derivatives (HADs) based on the 2018 EFSA opinion and any new data that have become available. Paper MUT/2021/11 provided a summary and discussion of the EFSA 2018 scientific opinion on the safety of HADs for use in food. Relevant literature studies published after 2018 were also described, including studies by Galli et al. (2021a,b) and Hu et al. (2021). Members were asked to consider whether they agreed with the EFSA 2018 conclusions and whether any of the EFSA conclusions would be affected by the results of the additional studies published since the EFSA 2018 opinion.
2.44 The Committee agreed that that overall, the available evidence indicated that emodin, aloe-emodin, and dantron are genotoxic in vitro, namely from Ames tests. Mixed results for in vitro genotoxicity had been reported in the literature. This was sometimes due to a lack of clarity on the preparation used for testing. Decolourised extracts (which were generally negative as they contain a far lower concentration of HADs), and whole extracts (which were positive as they contain greater concentrations of HADs). However, more information was needed to be confident that there was also genotoxicity in the mammalian cell assays, because i) the mouse lymphoma and micronucleus data summarised in the EFSA opinion were published in 1996 (since then, changes have been made to how genotoxicity is evaluated, for example to make sure excessive doses are not used), and ii) Müller et al. (1996) did not perform statistical evaluation of the data. Therefore, overall, it was not clear to the COM if the positive results in the mammalian cell assays were reflective of mutagenicity, or rather reflective of toxicity from the use of excessively high concentrations.
2.45 In terms of in vivo genotoxicity, one member questioned how much weight should be placed on negative mouse data published after 2018, as EFSA agreed that mice appear to be less sensitive than rats to the gastrointestinal effects caused by HADs. The Committee agreed that the studies published after 2018 are mostly negative in vivo data, which weaken the evidence that there is a genotoxic effect in vivo.
2.46 While EFSA concluded that results from the in vivo bone marrow micronucleus assay were irrelevant (due to insufficient bone marrow exposure), the COM noted that plasma analysis was conducted and the active compound was detected or quantified in plasma, indicating there was sufficient bone marrow exposure (albeit at a low level). COM further noted that a US National Toxicology Program (NTP) study included an assessment of plasma levels and micronucleus formation in rats and mice with acute intraperitoneal exposure (to ensure adequate systemic exposure) and these results were also negative. Therefore, the COM agreed that the negative results from the in vivo bone marrow micronucleus assay were valid and concluded that there is reasonable evidence that there is no genotoxic effect in vivo.
2.47 The COM considered that the carcinogenic effects of HADs, including those seen in the comet assay of colon cells, were caused by the high levels of irritation, inflammation, and diarrhoea. The 2-fold increase in tail moment (present at all dose levels) in colon cells under the comet assay was not caused by DNA reactivity, but rather an indirect mechanism involving ROS generation and/or topoisomerase II inhibition (mechanisms that were indicated from in vitro data). Since the Committee concluded that HADs do not show a genotoxicity mechanism, a new in vivo genotoxicity study would not be helpful.
2.48 The COM agreed that it should in theory be possible to establish a daily intake of HADs that does not give rise to health concerns using carcinogenicity data. However, more in vivo carcinogenicity data were needed to carry out dose response modelling and to identify a point of departure. The COM agreed that a specification for supplements regarding HADS contents would be useful for comparison against a potential Acceptable Daily Intake (ADI).
2.49 The FSA Secretariat agreed to provide an update to the COM in due course.
COM Horizon Scanning 2021
In this guide
In this guideForward look from the Chair
2.50 The Chair suggested two main areas of potential interest to the COM, which were genomics and next generation sequencing, and the use of genotoxicity markers in human biomonitoring. It was anticipated that in the next few years genomics and sequencing would be seen more in genotoxicity, including Duplex sequencing. There was a potential for this to support or even replace genotoxicity testing, particularly testing for gene mutation or point mutation. Developments in these areas may also provide an opportunity to gain more information from biomonitoring, occupational exposure or environmental exposure.
Presentation by Health and Safety Executive
2.51 Dr Lata Koshy gave a presentation on the work of the Health and Safety Executive (HSE) post the UK exit from the EU. HSE are involved in a number of activities within UK REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals), which includes identifying hazards, such as mutagenicity, and identifying substances of Very High Concern (SVHC). Most of the HSE work on Classification, Labelling and Packaging regulation relates to hazard identification for industrial chemicals. The HSE is also involved in the regulation of biocides and pesticides. Additionally, the HSE produces summaries for ministers and HSE opinions on the mandatory classification of substances and whether to align with EU opinion. The future work programme of the HSE is still being worked out post EU Exit and will be limited by resource and recruitment. HSE anticipated that it would complete the evaluation of two to three active substances per year. Evaluation of mutagenicity is a key part in determining whether an active substance will be given approval. Mutagenicity is also a key factor in the UK review of new and existing substances and import tolerance for pesticides. Due to the short timeline, it may be difficult consulting with COM, which has three meetings per year.
2.52 Some key differences for HSE since the UK exit from the EU is that the HSE has to act in isolation from EFSA and ECHA and from that peer review process. Its independence meant that it had to improve its own individual peer review process and has set up various expert groups and developed links with various other expert advisory groups. HSE may consult the COM in the future in relation to complex genotoxicity data sets and for advice in reviewing GHS for germ cell mutation category 1 and 2. The COM guidance documents, and expert advice will be useful to the HSE and its advice on specific areas, for example, on mode of action/threshold mode of genotoxic action and the use of QSARs.
Government assessors
2.53 Assessors from other Government Departments and agencies were asked for any horizon scanning topics they wished to highlight. VMD had an interest in biopharmaceutical molecules and their potential for mutagenicity. VMD were not aware of any guidance on how to assess the mutagenic potential, for example, of modified stem cells or monoclonal antibodies, particularly those sourced from different species (e.g. xenogeneic stem cells). VMD may seek the view of the COM of this area in the future. BEIS noted that it had set up its own expert scientific advisory groups following UK exit from the EU and that it would be seeking to develop links with secretariats for other expert advisory groups, such as the COM.
2.54 Members of the COM were asked to send in any thoughts on horizon scanning topics to the COM secretariat.
OECD
2.55 The COM was sent a consultation on a new draft Test Guideline on the mammalian erythrocyte Pig-a gene mutation assay. Members were requested to send any comments to the secretariat so that these could be collated and sent to the OECD.
OECD Draft Detailed Review Paper on the Miniaturised Versions of the Bacterial Reverse Gene Mutation Test
2.56 Members were requested to provide comments on an OECD Draft Detailed Review Paper (DRP) on the miniaturised Ames test (bacterial reverse gene mutation test) for collation by the National Coordinator at UK HSA. Assessors were requested to also send any comments which would be submitted separately.
2.57 It was noted that the DRP will not lead to a revision of the TG (TG471), but the aim of the review was to provide recommendations on the use of each of the mini-Ames tests proposed. From a UK perspective it was considered important to highlight and record any controversial points that were not in line with UK practice.
2.58 There was general agreement with the recommendations of the DRP. It was felt that until a robust validation process of the mini-Ames assays had been carried out, no further progress could be made in implementing the assays for regulatory testing. Further justification was requested, including better definition of what the assay is for, e.g., increasing output and reducing costs, incorporation of information relating to how laboratories were chosen to take part and whether there is a clear benefit of using mini-Ames assays above TG471. It was intended that a short written summary of the text submitted to OECD would be provided to COM members at the meeting in March 2022.
2021 Membership of the Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment
In this guide
In this guideChairman:
Dr David Lovell PhD BSc (Hons) FRSB CStat CBiol (until 30/04/2021).
Emeritus Reader in Medical Statistics at St George’s Medical School, University of London.
Professor Gareth Jenkins (from 01/05/2021).
Professor of Molecular Carcinogenesis, Faculty of Health, Medicine and Life Science, Swansea University.
Members:
Dr Carol Beevers
Broughton Nicotine Services.
Mr Amit Bhagwat
Lay Member.
Dr Stephen Dean (until 31/12/2021).
Imagen Therapeutics.
Professor Shareen Doak
Institute of Life Science, Swansea University Medical School.
Dr Paul Fowler
FSTox Consulting.
Professor David Harrison MD DSc FRCPath FRCPEd FRCSEd
Professor of Pathology, University of St Andrews.
Dr George Johnson
Associate Professor, Swansea University Medical School.
Ms Julia Kenny
GlaxoSmithKline.
Dr Ruth Morse (until 31/08/2021).
Senior Lecturer in Human & Clinical Genetics, University of the West of England, Bristol.
Dr Andrew Povey
Reader in Molecular Epidemiology, University of Manchester.
Mrs Madeleine Wang
Lay Member.
Secretariat
Dr Ovnair Sepai
PHE Scientific Secretary
Ms C Mulholland
FSA Scientific Secretary
Mrs N Blowfield
Administrative Secretary
Declaration of COM members interests during the period of this report 2021
In this guide
In this guideProfessor Gareth Jenkins COM Chair from 01/05/202.
|
Personal Interest |
Employer: Swansea University. |
|
Personal Interest |
Honorary Contract: Swansea Bay University Health Board. |
|
Personal Interest |
Membership: President of United Kingdom Environment Mutagen Society (UKEMS). Member: British Association for Cancer Research. Senior Editor Mutagenesis (OUP), Editorial Board (and former editor 2013-2015) Mutation Research (Elsevier). Health & Care Research Wales Grant panel (studentships) 2016 – present. |
|
Non-Personal Interest |
Grants: National Centre of Replacement, Refinement and Reduction of Animals in Research (NC3Rs) (2018-2022). Former NC3Rs grants (2012-2016 & 2010-2014) Former grants Health & Care Research Wales (2016-2020, 2014-2017. Unilever studentship 2014-2017. MRC/AstraZeneca PhD studentship (ITTP scheme) (2019-2023). Cancer Research Wales (2019-2023). BBSRC/Algae UK grant (2020-2022). |
Dr David Lovell PhD BSc (Hons) FRSB CStat CBiol COM Chair until 30/04/2021.
|
Personal Interest |
Pension: Pfizer. |
|
Personal Interest |
Membership: HESI GTTC (Committee member). Biometrics Society. British Toxicology Society (BTS). Genetics Society. Royal Society of Biology (RSB). Laboratory Animal Science Association (LASA). Royal Statistical Society {RSS). Statisticians in the Pharmaceutical Industry (PSI). United Kingdom Environment Mutagen Society, (UKEMS). UK National Centre of Replacement, Refinement and Reduction of Animals in Research (NC3Rs) – Board Member. MRC EMINENT Scientific Review Board. British Trust of Ornithologists (BTO). English Heritage. Liberty. Campaign of the Protection of Rural England (CPRE). Kew Gardens. Sandwich Bay Bird Observatory Trust (SBBOT). Chelsea Physic Garden. National Trust. |
|
Personal Interest |
Shareholder: National Grid plc. AstraZeneca (Spouse Shareholder). National Grid plc (Spouse Shareholder). |
|
Non-Personal Interest |
None. |
Dr Carol Beevers
|
Personal Interest |
Employee: Exponent International Ltd (up to 27 July 2021) Broughton Group (from 01 September 2021) |
|
Personal Interest |
Membership: HESI GTTC (workgroup member). OECD (workgroup member). IWGT (work group chair). United Kingdom Environmental Mutagen Society (UKEMS). |
|
Personal Interest |
Pension: Covance. Exponent. Broughton (from November 2021). |
|
Personal Interest |
Shareholder: ITM Power. NIO Inc. Blackberry. |
|
Non-Personal Interest |
None. |
Mr Amit Bhagwat
|
Personal Interest |
Owner and Shareholder: Research and Consulting Business. |
|
Personal Interest |
Trustee Myrovlytis Trust. Council on Nature Conservation and Countryside. |
|
Non-Personal Interest |
Bradford Teaching Hospitals NHS Foundation Trust - Public Governor (Rest of England & Wales). British Computer Society – the Chartered Institute for IT - Chair/Volunteer for Learned Events and Public Service Activities. NHS England subsidiary board on Mental Health Digital Programme – Public Member. Prescribed specialised services advisory group, DHSC – Public appointment. |
Dr Stephen Dean COM Member until 31/12/2021.
|
Personal Interest |
Employee: Imagen Therapeutics, From February 2020 (Equity Holder). |
|
Personal Interest |
Shareholder: Standard Life. |
|
Non-Personal Interest |
None. |
Professor Shareen Doak
|
Personal Interest |
Employee: Swansea University. |
|
Personal Interest |
Membership: United Kingdom Environmental Mutagen Society (UKEMS). Fellow of the Learned Society of Wales. British Association for Cancer Research (BACR). |
|
Non-Personal Interest |
Trustee: St David’s Medical Foundation (medical research & education charity). |
|
Non-Personal Interest |
PhD Studentship Grants: Unilever (2017 – 2020). AstraZeneca (2009 – 20160. Unilever (2010 -2017). |
|
Non-Personal Interest |
Research Grant: 2008 – 2010 Hoffman-LaRoche, Unilever. |
Dr Paul Fowler
|
Personal Interest |
Pension: Unilever (UK). Covance. Miscellaneous: De Montfort University – External Examiner. FSTox Consulting – Director. |
|
Personal Interest |
Membership: IGG (Chair). UKEMS (committee member). Roundtable of Toxicology Consultants (RTC). British Toxicology Society (BTS). EEMGS (Secretary). |
|
Non-Personal Interest |
None. |
Professor David Harrison
|
Personal Interest |
Employee: University of St Andrews, UK. NuCana plc, UK. |
|
Personal Interest |
Employee/Non-executive Director: ILC Therapeutics Ltd. Benenox Ltd, UK – Non-executive Director (unpaid). PathAlba Ltd – Director (unpaid) – dormant. |
|
Personal Interest |
Consultant: NHS Lothian – Honorary Consultant. |
|
Personal Interest |
Membership: None. |
|
Personal Interest |
Shareholder: VBL Ltd, UK. Ryboquin Ltd, UK. |
|
Personal Interest |
Miscellaneous: Cunningham Trust – Scientific Adviser. University of Edinburgh, UK – Honorary Professor. University of Glasgow, UK – Honorary Professor. University of Florida, Adjunct Professor. Viewbank Leuchars Ltd – Director (no salary). |
|
Non-Personal Interest |
Miscellaneous: iCAIRD research consortium – Director (unpaid role). Families First St Andrews (children’s charity) – Trustee (unpaid role). Visiopharm – Member, Scientific Advisory Board. Royal College of Pathologists – Fellow. Royal College of Physicians of Edinburgh – Fellow. Royal College of Surgeons of Edinburgh – Fellow. UK Committee on Mutagenicity – Member. Scottish Government A1 Leadership Circle – Member. EU Horizon 2020, Partner in KATY Award, grant support. Innovate UK/UKRI – Director of iCAIRD. |
Dr George Johnson
|
Personal Interest |
Consultancy: Fermenich, Cefic, American Chemistry Council, Teva, Greenberg Traurig llp, Osler, Hoskin & Harcourt llp, Janssen, Merck. |
|
Personal Interest |
Pension: USS (university superannuation scheme). |
|
Personal Interest |
Director: GTox ltd. |
|
Personal Interest |
Membership: United Kingdom Environmental Mutagen Society (UKEMS). HESI (committee member). President of the European Environmental Mutagenesis and Genomics Society (EEMGS) 2019-2021. EMA expert member. IWGT, expert member. ICEM, committee member. |
|
Non-Personal Interest |
Relevant grant funding: GSK, post-doctoral research funding – 2021-2022 nitrosamine research. SCIENSANO. MYCX-IT. 2020-ongoing. EMA. funding through Fraunhofer item. 2022-2023. HESI. fast fund. msc tuition fees 2022. |
Ms Julia Kenny
|
Personal Interests |
Employee: GlaxoSmithKline. |
|
Personal Interests |
Pension: GlaxoSmithKline. |
|
Personal Interests |
Shareholder: GlaxoSmithKline. |
|
Personal Interests |
Membership: UK Environmental Mutagen Society (UKEMS). |
|
Non-Personal Interest |
None. |
Dr Ruth Morse COM Member until 31/04/2021.
|
Personal Interest |
Member: United Kingdom Environmental Mutagen Society. British Society of Toxicology. Genetics Society. |
|
Non-Personal Interest |
Miscellaneous: Medical Research Council with AstraZeneca. (ITTP programme) - PhD studentship collaborative grant 2015-2020. Petroleum Technology Fund, Nigeria – PhD. Studentship 2016-2020. |
Dr Andrew Povey
|
Personal Interest |
Shareholder: Lloyds, Standard Life, Halifax, Santander (Partner Shareholder), Norwich Union (Partner Shareholder), Roadchef Topco Ltd (Partner Shareholder). |
|
Personal Interest |
Miscellaneous: European Crop Protection Agency – Part of consortium recently awarded grant on exposure assessment. |
|
Personal Interest |
Membership: UK Molecular Epidemiology Group (UK-MEG). UK Environmental Mutagen Society (UKEMS). American Association for Cancer Research (AACR) Molecular Epidemiology Group (MEG). British Association for Cancer Research (BACR). |
|
Non-Personal Interest |
Miscellaneous: RTZ – Departmental Research Grant. Manchester University – Research equipment bought using departmental funds from consultancies with industry and other bodies. |
Ms Madeleine Wang
|
Personal Interest |
None. |
|
Non-Personal Interest |
None. |
Committee on the Carcinogenicity of Chemicals in Food, Consumer Products and the Environment 2021
In this guide
In this guide
Head and shoulders image of Professor David Harrison in front of a grey background wearing a white shirt with a blue and yellow striped tie.
The Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment (COC) evaluates chemicals for their potential to cause cancer in humans at the request of UK Government Departments and Agencies.
The membership of the Committee, agendas and minutes of meetings, and statements are all published on the internet (Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment).
This year’s meetings continued in virtual form, and I am grateful to Members, Secretariat and other contributors for ensuring that the work continues, despite the challenges this has brought.
The Committee have this year started considerations around relevance and reliability of evidence, a piece of work being jointly considered with COM and COT and this will continue into 2022, and aligned with work being undertaken by the FSA Science Council on third party evidence.
We have continued our review and update of the COC guidelines, undertook our annual horizon scanning, and continued the discussion on modification of risk of developing clinical cancer by chemicals as part of our efforts to review the conceptual framework we use for assessment and advice. This last topic will take time to come to fruition but crucially relies on Members being aware of, and integrating, new results from emerging technologies. We will continue to work on developing our work in this area in the coming years to ensure our guidance to Government Departments and Agencies is relevant to the challenges we face.
Professor David Harrison
MD DSc FRCPath FRCPEd FRCSEd
COC Evaluations 2021
In this guide
In this guideHuman Biomonitoring for EU and Development of Human Biomonitoring Guidance Values in the HBM4EU project
3.1 A presentation was given by Dr Sepai, Public Health England (PHE), at the COC meeting on 11th March 2021 and the COT meeting on March 23rd, 2021, with a supporting paper ‘Development of Human Biomonitoring Guidance Values in the HBM4EU biomonitoring project’.
3.2 Human biomonitoring (HBM) programmes can provide essential information for identifying population exposures to chemicals of concern that can be assessed with regards to potential health risks against derived guidance values (GVs) in specific population subgroups or areas. These can be important complements to the conventional sources of information for regulatory chemical risk assessments and for supporting public and occupational health protection policies.
3.3 There is currently a diversity in the derivation of health-based guidance values for both the general population and for occupational exposures. Dr Sepai outlined the methodology for the derivation of human biomonitoring guidance values (HBM-GVs) by the European Human Biomonitoring Initiative, referred to as HBM4EU. This is a project involving 30 countries, the European Environment Agency and the European Commission, co-funded under Horizon 2020. The UK has been involved in the project with PHE leading the UK input. The initiative is designed to develop a harmonised and systematic strategy for the derivation of HBM-GVs.
3.4 Importantly, the HBM4EU strategy is based on current practices for deriving health-based assessment values based on internal exposure, which will supplement those already derived relating to external exposure measurements. The key schemes on which the HBM-GV derivation methodology is based are those already existing from the German Human Biomonitoring Commission, Summit Toxicology and the French Agency for Food, Environmental and Occupational Health & Safety. Members of the COC and COT were asked to consider whether the derived HBM-GVs could be used for risk assessment purposes and if the HBM-GVs would be accepted by the UK.
3.5 It was agreed, in principle, by members of both Committees that the framework was a robust and scientifically valid way to determine HBM-GVs but offered suggestions to make some components of the process more explicitly stated, including the impact of data availability (for example, toxicokinetic data) on the estimated level of confidence associated with each HBM-GV. It was accepted that the estimated level of confidence would vary on a case-by-case basis, depending on available data, which should be reflected in the use of the HBM-GV in different tiers for risk assessment purposes. As the values are able to be applied to any population, the absence of UK-specific population data was not considered an issue for derivation, with the caveat that the critical endpoint on which the HBM-GV was derived is appropriate for the UK population. However, members considered that UK-specific data would be required before the HBM-GVs could be used for risk assessment purposes in the UK.
3.6 The COT commented that the HBM-GV’s would need to be validated from a toxicological perspective (see paragraph 1.78). It was also suggested that refinements in exposure assessment could be achieved through the collection of environmental data (in collaboration with the Environment Agency or Defra) and through the inclusion of all routes of exposure, including dermal. Members agreed that going forward, the use of HBM-GVs in risk assessment could be particularly helpful to the FSA and that the Committee was happy to look at future case studies and offer their perspective. If endorsement of individual values was needed, the Committee would have to perform a detailed evaluation to offer their opinion.
Modification of Cancer Risk
3.7 COC had expressed its aspiration in the preceding years to move away from traditional risk assessment approaches for potential carcinogens, to a more holistic approach encompassing consideration of the modifying effects of chemicals on all stages of cancer development. This has been reinforced by increasing concern over the reliability and applicability of the rodent two-year bioassay in predicting chemical carcinogenicity relevant to humans. In addition, consideration had also been made of combining two guidance statements covering hazard identification and characterisation (G03), and alternatives to the two-year bioassay (G07) to a combined document on considering modification of cancer risk using a weight of evidence-based approach.
3.8 The COC discussed this further in 2021, in the main Committee and as a sub-group discussion. It was agreed that there was currently insufficient information available on all aspects of cancer development and the potential modification of these events by chemicals to facilitate its use by risk assessors. Therefore, distinct COC guidance could not be developed at this point, but two guidance statements G03 and G07 should be updated (see 3.26 below). A paper capturing these thoughts was published in Toxicology Research by two members, (Harrison & Doe (2021) The modification of cancer risk by chemicals. Toxicology Research, 10(4), 800-809). This covered many of the aspects discussed by the Committee and it was agreed the topic would not be progressed to a separate published COC document.
FSA Science Council Draft Principles and Guidelines on Third Party Evidence
3.9 The COC was presented the draft set of principles and guidelines on third party and uncommissioned evidence that had been prepared by the FSA Science Council to support consideration of such evidence and provide transparency on the ways in which evidence submitted in a non-standard way would be assessed.
3.10 The COC made some suggestions for clarity in terms of the audience for the principles and guidelines and to be clear on the meaning of the wording on data cleaning.
3.11 The document has subsequently been finalised by the FSA Science Council. See Rapid Evidence Review on the Critical Appraisal of Third-Party Evidence (food.gov.uk) for further details.
Terms of reference for the Office for Product Safety and Standards (OPSS) Scientific Advisory Group on Chemical Safety of Non-Food and Non-Medicinal Consumer Products (SAG-CS)
3.12 The terms of reference for the Office for Product Safety and Standards (OPSS) Scientific Advisory Group on Chemical Safety of Non-Food and Non-Medicinal Consumer Products (SAG-CS) was presented to the COC for awareness of this group. The COC fed back the suggestion of having lay representation on the group in the future.
Presentation by Dr Steve Dean “In vitro high content screening using patient-derived cell models”
3.13 The presentation by Dr Steve Dean, Imagen, described a personalised treatment for cancer that evaluates potential drug therapies using patient derived cell models. The PredictRx assay utilises a biopsy from patients to derive cells that are screened against 60 drugs to determine sensitivity of the tumour cells. They report a good prediction of clinical response with an 89% positive predictive value and 99% negative predictive value for those currently tested. Due to the low number and heterogeneous nature of the tumours, between 3 and 5 needle biopsies are usually taken which are pooled. The results therefore represent an average of the responses of the different tumour cells.
3.14 Since 2019, with informed consent, the patient-derived cells have been stored in a biobank and a searchable database has been established. The biobank has a range of solid tumour types and is being expanded to include haematological tumours. As with primary cell lines, patient-derived cell models generally have a limited life span, and to ensure that the cell models do not diverge from the original, a limited number of passages are allowed. The biobank and database are a key resource for the evaluation of new drug candidates at all stages of development, including the potential to enhance Phase I II and III clinical trials.
3.15 The biobank and database are also seen as a potentially interesting resource for cancer research to help gain an understanding of carcinogenicity and mutagenicity. Advantages include high throughput analysis of a range of endpoints including cytotoxicity and apoptosis, cell cycle, DNA damage & repair, morphological and phenotypic changes, cell stress and inflammation, cell signalling and transcription factors and drug internalisation. Importantly, the cell models are reliable pre-clinical models with a traceable origin and are accompanied by patient histories.
3.16 Following the presentation, the COC noted that this was potentially a good example of how in vitro methodology may allow risk assessors to steer away from the use of traditional in vivo study data and allow better understanding of mechanisms in humans. The stability of the cell models was questioned as this was seen as crucial to ensure that the models continued to represent the patient. As this could be different for each model, whilst this is being evaluated, the models are currently limited to 15-20 passages. It was recognised that validation will be key to getting clinical acceptance as a diagnostic tool and acceptance of findings within regulatory submissions.
3.17 The translatability of the approach, particularly the data, to establish mechanistic rather than response data was also raised. This had been attempted successfully for a metabolic syndrome and was believed to be applicable more widely to non-cancer endpoints. Artificial Intelligence platforms may play a key role in interpreting mechanistic data. Benefits of the use of the approach to assess risk included the high throughput nature, availability of detailed genotypic and phenotypic parameters and a response pathway analysis.
COC Joint Ongoing Topics 2021
In this guide
In this guideRelevance and Reliability of Evidence
3.18 The topic of ‘biological relevance and statistical significance’ has been raised as an area of interest during Committee horizon scanning activities for a number of years. A scoping paper was presented at the Joint COC/COM meeting in November 2020 also attended by some COT members, which outlined some of the more relevant and significant work that has been published on this issue in recent years. It was agreed that the general public would benefit from guidance that provided clarity on how the expert Committees evaluate data with respect to consideration of biological relevance and statistical significance.
3.19 A document providing a brief outline of the Committee evaluation process focussing on the relevance and reliability of data was drafted and discussed by COC and COT in 2021. During the COC and COT discussions it was proposed that two documents be developed. One aimed at the lay audience about the process used by the Committees to evaluate evidence and reach conclusions, which could possibly be presented on the website rather than formally as a statement. A second document aimed at a more informed audience on statistical significance testing and consideration of biological relevance, for which the current draft would be the basis.
3.20 This topic will be discussed further, including by COM.
COC Horizon Scanning 2021
In this guide
In this guide3.21 The COC undertakes horizon scanning exercises at regular intervals with the aim of identifying new and emerging issues which have potential to impact on public health.
3.22 At the end of discussion in 2021, it was agreed that the priority topics were:
- Maintain a watching brief on factors affecting cancer susceptibility including shift work, stress and other lifestyle factors and how that might affect assessment of chemicals and carcinogenicity.
- Consider an update to guidance on assessment of nanomaterials, possibly as a joint activity across COC, COM and COT.
- Gain awareness of the potential effects of antibiotics and antivirals on the microbiome.
- Consider a joint discussion with COM on thresholds for in vivo mutagens and whether there is new information subsequent to the 2010 COM opinion.
- Endocrine disruption and the link with carcinogenicity, acknowledging that endocrine disruption is also within the COT remit.
- Impact of chemicals on potential for metastasis or progression of cancer, in particular with respect to the tumour microenvironment.
- Communication of cancer risk and how COC should be involved with this, especially with the move away from a yes/no decision on whether a substance is a carcinogen, and ensuring consistency in describing risks, possibly starting with a landscape review of terminology across a number of Committees (FSA and UKHSA) and led by Lay Members.
- Ensuring appropriate considerations are made to acknowledging diversity in the population especially where there might be differences in risk between different groups.
3.23 The Committee continues to have a standing agenda item for each meeting on horizon scanning topics and to update the COC on upcoming topics for UK and international scientific advisory groups.
COC Working Groups 2021
In this guide
In this guideCOT/COC subgroup on the synthesis and integration of epidemiological and toxicological evidence in risk assessment
3.24 The COT and COC set up a subgroup to review the approaches to synthesising epidemiological and toxicological evidence that are used in chemical risk assessments. More information is provided in the COT section 1.150-1.157
COC Guidance Statements 2021
In this guide
In this guide3.25 The Committee continued to develop the guidance statement series during 2021. This included finalising revisions to the cancer risk characterisation methods (G06) statement.
3.26 Updates to the guidance on hazard identification and characterisation (G03), the use of biomarkers in carcinogenic risk assessment (G04) and alternatives to the two-year bioassay (G07) are ongoing and these are expected to be finalised in 2022.
2021 Membership of the Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment
In this guide
In this guideChairman:
Professor David Harrison MD DSc FRCPath FRCPEd FRCSEd
Professor of Pathology, University of St Andrews.
Members:
Mr Derek Bodey MA
Public Interest Representative.
Dr Gill Clare BSc PhD
Independent Consultant in Genetic Toxicology.
Dr Meera Cush
Managing Consultant (Toxicologist), Ramboll.
Dr Ruth Dempsey
Consultant: RD Science Speaks Consultancy, Sàrl.
Dr John Doe PhD
Research Fellow, Liverpool John Moore’s University.
Dr Richard Haworth MA VetMB DPhil FRCPath DipECVP DABT
Head of Pathology UK, GlaxoSmithKline.
Dr Ray Kemp BA MSc PhD MRTPI SIRM
Public Interest Representative.
Dr David Lovell PhD BSc (Hons) FRSB CStat CBiol (until 30th April 2021).
Emeritus Reader in Medical Statistics at St George’s Medical School, University of London.
Professor Gareth Jenkins (from 1st May 2021).
Professor of Molecular Carcinogenesis, Faculty of Health, Medicine and Life Science, Swansea University.
Professor Neil Pearce BSc DipSci DipORS PhD DSc FRSNZ FMedSci FFPH
Professor of Epidemiology and Biostatistics, London School of Hygiene and Tropical Medicine.
Dr Lesley Rushton OBE BA MSc PhD CStat HonFFOM
Emeritus Reader in Occupational Epidemiology, Imperial College London.
Dr Lesley Stanley
Consultant in Investigative Toxicology.
Professor Heather Wallace BSc(Hons) PhD FRCPath FBTS FRSC FRSB FBPS ERT
Professor in Biochemical Pharmacology and Toxicology, University of Aberdeen.
Secretariat
Miss B Gadeberg BSc(Hons) MSc
PHE Scientific Secretary to 30 Sept 2021. UKHSA Scientific Secretary from 1st October 2021.
Ms Catherine Mulholland BSc (Hons), ERT
Scientific Secretary
Dr D Gott BSc(Hons) PhD
FSA Scientific Secretary
Mrs N Blowfield
Administrative Secretary
Declaration of COC members interests during the period of this report 2021
In this guide
In this guideProfessor David Harrison
|
Personal Interest |
Employee: University of St Andrews UK, NuCana plc UK. |
|
Personal Interest |
Employee/Non-executive Director: ILC Therapeutics Ltd. Benenox Ltd, UK – Non-executive Director (unpaid). PathAlba Ltd – Director (unpaid) – dormant. |
|
Personal Interest |
Consultant: NHS Lothian – Honorary Consultant. |
|
Personal Interest |
Shareholder VBL Ltd UK, Ryboquin Ltd UK. |
|
Personal Interest |
Miscellaneous: Cunningham Trust – Scientific Adviser. University of Edinburgh, UK – Honorary Professor. University of Glasgow, UK – Honorary Professor. University of Florida, Adjunct Professor. Viewbank Leuchars Ltd – Director (no salary). |
|
Non Personal Interest |
Miscellaneous: iCAIRD research consortium – Director (unpaid role). Families First St Andrews (children’s charity) – Trustee. (unpaid role). Visiopharm – Member, Scientific Advisory Board. Royal College of Pathologists – Fellow. Royal College of Physicians of Edinburgh – Fellow. Royal College of Surgeons of Edinburgh – Fellow. UK Committee on Mutagenicity – Member. Scottish Government A1 Leadership Circle – Member. EU Horizon 2020, Partner in KATY Award, Grant Support. Innovate UK/UKRI – Director of iCAIRD. |
Mr Derek Bodey
|
Personal Interest |
None |
|
Non Personal Interest |
None |
Dr Gill Clare
|
Personal Interest |
Pension: Shell Research Ltd, AstraZeneca. |
|
Personal Interest |
Shareholder: AstraZeneca, Diageo, Marks and Spencer. |
|
Personal Interest |
Consultant: Covance. |
|
Personal Interest |
Miscellaneous: OPSS Register of Specialists from December 2020. OPSS Scientific Advisory Group from March 2021. Food Standards Agency (FSA) Joint Expert Group on Food Contact Materials (FCM-JEG) from May 2019. HSE REACH Independent Scientific Expert Pool from June 2021. Member of joint COT and COC Synthesis and Integration of Epidemiological and Toxicological Evidence sub-group, 2019 – 2021. |
|
Non Personal Interest |
Membership: United Kingdom Environmental Mutagen Society (UKEMS). |
|
Non-Personal Interest |
None. |
Dr Meera Cush
|
Personal Interest |
Employee: University of Surrey – Visiting Lecturer. |
|
Personal Interest |
Membership: Royal Society of Biology. |
|
Non-Personal Interest |
None. |
Dr Ruth Dempsey
|
Personal Interest |
Shareholder: RD Science Speaks Consultancy, Sarl (Shareholder and director). |
|
Personal Interest |
Membership: British Toxicology Society. Swiss society of Toxicology. Royal society of Biology. |
|
Personal Interest |
Pension: Philip Morris International. |
|
Personal Interest |
Consultant: Philip Morris International, doTERRA Europe. |
|
Non-Personal Interest |
None. |
Dr John Doe PhD
|
Personal Interest |
Associate: Regulatory Science Associates Ltd. |
|
Personal Interest |
Membership: British Toxicology Society (BTS). |
|
Personal Interest |
Pension: Syngenta |
|
Personal Interest |
Consultant: ECETOC, Syngenta, Covance. |
|
Personal Interest |
Miscellaneous: Liverpool John Moores University (Honorary Research Fellow). |
|
Non-Personal Interest |
None. |
Dr Richard Haworth
|
Personal Interest |
Employee: GlaxoSmithKline. |
|
Personal Interest |
Membership: British Society of Toxicological Pathology. |
|
Personal Interest |
Shareholder: GlaxoSmithKline. Royal Dutch Shell (Spouse Shareholder). United Utilities (Spouse Shareholder). |
|
Non-Personal Interest |
None. |
Dr Ray Kemp
|
Personal Interest |
Director: Rhodes-Kemp Law Ltd. |
|
Personal Interest |
Member: Royal Town Planning Institute Specialist. Committee on Medical Aspects of Radiation in the Environment (COMARE). Institute of Risk Management. Committee on Radioactive Waste Management (CoRWM). |
|
Personal Interest |
Non-Executive Director: Dept of Business, Energy and Industrial Strategy (BEIS). |
|
Personal Interest |
Independent Expert: International Atomic Energy Agency – Mission to Fukushima Prefecture. |
|
Personal Interest |
Independent Expert: Office for Rail and Road. |
|
Non-Personal Interest |
None. |
Dr David Lovell PhD BSc (Hons) FRSB CStat CBiol COC Member until 30th April 2021.
|
Personal Interest |
Pension: Pfizer. |
|
Personal Interest |
Membership: Biometrics Society. British Toxicology Society (BTS). Genetics Society. Royal Society of Biology (RSB). Laboratory Animal Science Association (LASA). Royal Statistical Society (RSS). Statisticians in the Pharmaceutical Industry (PSI). United Kingdom Environment Mutagen Society (UKEMS). UK National Centre of Replacement, Refinement and Reduction of Animals in Research (NC3Rs) – Board Member. MRC EMINENT Scientific Review Board. British Trust of Ornithologists (BTO). English Heritage. Liberty. Campaign of the Protection of Rural England (CPRE). Kew Gardens. Sandwich Bay Bird Observatory Trust (SBBOT). Chelsea Physic Garden. National Trust. HESI GTTC (Committee member). |
|
Personal Interest |
Shareholder: National Grid plc. AstraZeneca (Spouse Shareholder). National Grid plc (Spouse Shareholder). |
|
Non-Personal Interest |
None. |
Professor Gareth Jenkins COC Member from1st May 2021.
|
Personal Interest |
Employer: Swansea University. |
|
Personal Interest |
Membership: President: of United Kingdom Environment Mutagen Society (UKEMS). Member: British Association for Cancer Research. |
|
Personal Interest |
Honorary Contract: Swansea Bay University Health board. |
|
Personal Interest |
Senior Editor: Mutagenesis (OUP), Editorial Board (and former editor 2013-2015) Mutation Research (Elsevier). Health & Care Research Wales Grant panel (studentships) 2016-present. |
|
Non-Personal Interest |
Grants: National Centre of Replacement, Refinement and Reduction of Animals in Research (NC3Rs) (2018-2022). Former NC3Rs grants (2012-2016 & 2010-2014). Former grants Health & Care Research Wales (2016-2020, 2014-2017. Unilever studentship 2014-2017. MRC/AstraZeneca PhD studentship (ITTP scheme) (2019-2023). Cancer Research Wales (2019-2023). BBSRC/Algae UK grant (2020-2022) |
Professor Neil Pearce
|
Personal Interest |
None. |
|
Non-Personal Interest |
None. |
Dr Lesley Rushton OBE BA MSc PhD Cstat HonFFOM
|
Personal Interest |
Member: Industrial Injuries Advisory Council – Chair. |
|
Non-Personal Interest |
Miscellaneous: IEH Consultancy Ltd – Research Support. |
Dr Lesley Stanley
|
Personal Interest |
Self-employed: Dr Lesley Stanley, Consultant in Investigative Toxicology. |
|
Personal Interest |
Membership: European Registered Toxicologist (ERT.) Fellow of the British Toxicology Society (FBTS). Advisory Committee on Novel Foods and Processes (ACNFP). |
|
Personal Interest |
Consultancy: School of Medicine, University of Dundee (2020 to date). Details of previous consultancy contracts available upon request. |
|
Personal Interest |
Expert Appointments: REACH Independent Scientific Expert Pool. OPSS Register of Experts. |
|
Personal Interest |
Honorary Appointment: Associate, School of Life Sciences, Edinburgh Napier University (Non-Stipendiary). |
|
Personal Interest |
Investments: Investment Portfolio managed by Quilter Cheviot (joint with spouse). FundsNetwork Stocks and Shares ISA. Aviva Personal Pension Plan. |
|
Personal Interest |
Ministry and Charities: Ordained Local Minister, Church of Scotland (non-stipendiary). Honorary Chaplain, University of Stirling (non-stipendiary). Supporter, Christian Aid “In Their Lifetime” programme and International Justice Mission. |
|
Non-Personal Interest |
None. |
Professor Heather Wallace BSc Hons PhD FRCPath FBTS FRSC FRSB ERT
|
Personal Interest |
Shareholder: Bank Santander SA, BT Group, NovaBiotics, Aviva. |
|
Personal Interest |
Membership: EUROTOX – Past President. British Toxicological Society (BTS). Medical Research Scotland – Chair and Trustee. Paediatric Medicines Expert Advisory Group – MHRA. Herbal Medicines Advisory Committee – MHRA. |
|
Personal Interest |
Miscellaneous: EFSA – CONTAM Panel. Cell ProTx – Director. |
|
Non-Personal Interest |
None. |
Annex 1 - Terms of Reference
In this guide
In this guideTo advise at the request of:
Food Standards Agency
Food Standards Scotland
Public Health England
Department of Health and Social Care
Other Government Departments and Agencies and those of the UK devolved Administrations.
1. To assess and advise on the toxic risk to man of substances which are:
a) used or proposed to be used as food additives or used in such a way that they might contaminate food through their use or natural occurrence in agriculture, including horticulture and veterinary practice or in the distribution, storage, preparation, processing or packaging of food.
b) used or proposed to be used or manufactured or produced in industry, agriculture, food storage or any other workplace.
c) used or proposed to be used as household goods or toilet goods and preparations.
d) used or proposed to be used as drugs, when advice is requested by the Medicines and Healthcare products Regulatory Agency.
e) used or proposed to be used or disposed of in such a way as to result in pollution of the environment.
2. To advise on important general principles or new scientific discoveries in connection with toxic risks, to co-ordinate with other bodies concerned with the assessment of toxic risks and to present recommendations for toxicity testing.
Annex 2 - Code of Conduct for members of the COC/COM/COT
In this guide
In this guidePublic service values
Members of the COC/COM/COT (hereafter referred to as “the Committee”) must at all times:
-
observe the highest standards of impartiality, integrity and objectivity in relation to the advice they provide and to the management of their Committee.
-
be accountable, through the Chair of the Food Standards Agency and the Chief Medical Officers, to Ministers, Parliament and the public for its activities and for the standard of advice it provides.
-
in accordance with Government policy on openness, fully comply with the Freedom of Information Act 2000.
The Ministers of the sponsoring departments are answerable to Parliament for the policies and performance of the Committee, including the policy framework within which it operates.
Standards in Public Life
Members are expected to:
-
comply with this Code, and ensure they understand their duties, rights and responsibilities, and that they are familiar with the function and role of their Committee and any relevant statements of Government policy. If necessary members should consider undertaking relevant training to assist them in carrying out their role.
-
not misuse information gained in the course of their public service for personal gain or for political purpose, nor seek to use the opportunity of public service to promote their private interests or those of connected persons, firms, businesses or other organisations.
-
not hold any paid or high profile unpaid posts in a political party, and not engage in specific political activities on matters directly affecting the work of the Committee. When engaging in other political activities, Committee members should be conscious of their public role and exercise proper discretion. These restrictions do not apply to MPs (in those cases where MPs are eligible to be appointed), to local councillors, or to Peers in relation to their conduct in the House of Lords.
-
follow the Seven Principles of Public Life set out by the Committee on Standards in Public Life Committee on Standards in Public Life - GOV.UK (www.gov.uk)
Selflessness
Holders of public office should take decisions solely in terms of the public interest. They should not do so in order to gain financial or other material benefits for themselves, their family, or their friends.
Integrity
Holders of public office should not place themselves under any financial or other obligation to outside individuals or organisations that might influence them in the performance of their official duties.
Objectivity
In carrying out public business, including making public appointments, awarding contracts, or recommending individuals for rewards and benefits, holders of public office should make choices on merit.
Accountability
Holders of public office are accountable for their decisions and actions to the public and must submit themselves to whatever scrutiny is appropriate to their office.
Openness
Holders of public office should be as open as possible about all the decisions and actions that they take. They should give reasons for their decisions and restrict information only when the wider public interest clearly demands.
Honesty
Holders of public office have a duty to declare any private interests relating to their public duties and to take steps to resolve any conflicts arising in a way that protects the public interests.
Leadership
Holders of public office should promote and support these principles by leadership and example.
These principles apply to all aspects of public life. The Committee has set them out here for the benefit of all who serve the public in any way.
Role of Members
Members have collective responsibility for the operation of their Committee. Members are appointed as individuals to fulfil the role of their respective Committees, not as representatives of their particular profession, employer or interest group and have a duty to act in the public interest. Members are appointed on a personal basis, even when they are members of stakeholder groups and organisations. If a member declares an organisation’s view rather than a personal view they should make it clear at the time of declaring that view.
Members must:
-
engage fully in collective consideration of the issues, taking account of the full range of relevant factors, including any guidance issued by the Food Standards Agency, Health Protection Agency and the Department of Health.
-
undertake on appointment to comply with the Code of Practice for Scientific Advisory Committees.
-
not divulge any commercially sensitive information, pre-publication or unpublished research data provided to the Committee.
-
agree an annual report.
-
ensure that an appropriate response is provided to complaints and other correspondence, if necessary with reference to the sponsor department.
-
ensure that the Committee(s) does not exceed its powers or functions.
A member’s role on the Committee should not be limited by the expertise or viewpoint she or he was asked to bring to it. Any statement/report belongs to the whole Committee. Members should regard themselves free to question and comment on the information provided or the views expressed by any of the other members, even though the views or information provided do not relate to their own area of expertise.
If members believe the committee’s method of working is not rigorous or thorough enough, they have the right to ask that any remaining concerns they have be put on the record. Individual members should inform the Chair (or the Secretariat on his or her behalf) if they are invited to speak in public in their capacity as a Committee member. Communications between members and the Food Standards Agency (FSA) Board, CMOs and/or Ministers will generally be through the Chair except where the Chair has agreed that an individual member should act on its behalf. Nevertheless, any member has the right of access to the FSA Board and/or the CMO on any matter that he or she believes raises important issues relating to his or her duties as a Committee member. In such cases the agreement of the rest of the Committee should normally be sought.
Committee appointments can be terminated early by either party, by giving 3 months’ notice, in writing. Should the Committee be disbanded before the end of the period of appointment, appointments will terminate on dissolution.
In the event that a member is found guilty of grave misconduct their appointment will be terminated immediately, in the case of the COT by the Chair of the FSA. The Department of Health has delegated the powers for appointments to the COC and COM to the NHS Appointments Commission and it will terminate appointments in consultation with the PHE/DH.
Role of the Chair
The Chair has particular responsibility for providing effective leadership on the issues above. In addition, the Chair is responsible for:
-
ensuring that the Committee meets at appropriate intervals,
-
ensuring that the minutes of meetings accurately reflect proceedings and any reports to the FSA Board and/or Ministers accurately record the decisions taken,
-
ensuring that where appropriate, the views of individual members have been recorded,
-
representing the views of the Committee to the general public,
-
ensuring that new members are briefed on appointment (and their training needs considered), and providing an assessment of their performance, on an annual basis or when members are considered for re-appointment to the Committee or for appointment to the board of some other public body,
-
providing urgent advice to the FSA and HPA on issues within the remit of the Committee, in liaison with the Secretariat.
Role of the Deputy Chair
The Deputy Chair will assume the role of the Chair as described above if the Chair is not available.
Role of the Secretariat
The primary function of the Secretariat is to facilitate the business of the Committee. This includes supporting the Committee by arranging its meetings, assembling and analysing information, and recording conclusions. An important task is ensuring that proceedings of the Committee are properly documented and recorded. Minutes of all Committee meetings will be taken. These will accurately reflect the proceedings and discussions that take place and will be recorded on a non-attributable basis except where the views of one or more individual members need recording (for example, when declaring an interest).
The Secretariat is also a source of advice and guidance to members on procedures and processes. The Secretariat is drawn from staff of the Food Standards Agency and Public Health England. However, it is the responsibility of the Secretariat to be an impartial and disinterested reporter and at all times to respect the Committee’s independent role. The Secretariat is required to guard against introducing bias during the preparation of papers, during meetings, or in the reporting of the Committee’s deliberations. Current contact details for each of the Secretariats are shown on the back page of this report.
Role of the Assessor
Meetings of the Committee (and working groups) may be attended by Assessors. The Assessors are nominated by, and drawn from, the Agencies and Departments that sponsor the Committee, receive its advice, or have other relevant policy interests. Assessors are not members of the Committee and do not participate in Committee business in the manner of members.
The role of an Assessor is to keep their parent Department or Agency informed about the Committee’s work and act as a conduit for the exchange of information. They do this by:
-
advising the Committee on relevant policy developments and the implications of Committee proposals,
-
informing the Committee work through the provision of information,
-
being informed by the Committee on matters of mutual interest,
-
sharing with the Secretariat the responsibility of ensuring that information is not needlessly withheld from the Committee. Assessors should make the Committee aware of the existence of any information that has been withheld from the Committee on the basis that it is exempt from disclosure under Freedom of Information legislation unless that legislation provides a basis for not doing so,
-
ensuring that their parent Department or Agency is promptly informed of any matters which may require a response from Government.
Role of other Officials, Invited Experts and Contractors
Officials from Government Departments (not departmental assessors), Regulatory Agencies and Devolved Administrations may be called upon to advise the Committee on relevant developments in order to help the Committee formulate its advice. Invited experts and contractors may also bring particular technical expertise, which may be requested by the Committee on some occasions. In the event of an official, invited expert or contractor not being able to attend written submissions may be sent via the Secretariat.
Role of Observers
Members of the public and other interested parties may attend meetings as observers. However, they should not attempt to participate in Committee discussions. If an interested party wishes to provide information relevant to a topic for consideration by the Committee, they should be submitted in writing to the Secretariat at least seven (7) working days before the meeting. The Secretariat will discuss with the Chair the most appropriate way to present the information to the committee and the Chair's decision will be final.
Observers who have submitted information in advance of the meeting may be invited to provide further explanation or to make brief comments at the discretion of the Chair. Observers and/or organisations must not interfere in the work of the Secretariat or input from invited experts, contractors, officials from Government Departments and Agencies in any way which, in the view of the Chair, constitutes harassment and/or might hinder the work of the Committee. Observers and/or organisations must allow other observers and other interested parties to attend items free from interference before, during and after a meeting.
Observers and/or organisations are required to respect the work of the Committee. The Committee's discussions represent the development of its view and any comments made in developing the agreed Committee view should not be attributed to individuals. Where a subject will be considered over several meetings, observers are asked to maintain the confidentiality of the discussion until an agreed Committee opinion is finalised. The Committee's conclusions are not finalised until completion of any necessary consultation and publication of a statement or report.
Under no circumstances will Observers be permitted to record Committee proceedings, on the basis that this might inhibit free discussion. The published minutes of the meeting would provide a record of the proceedings.
Failure to observe this code of conduct may lead to exclusion of individual observers and/or organisations from meetings of the Committee.
All observers and/or organisations are requested to read follow the Committees Openness policy (Annex 3).
Declaration of Members’ Interests
Definitions
In this Code, ‘the industry’ means:
-
Companies, partnerships or individuals who are involved with the production, manufacture, sale or supply of products subject to the following legislation.
General Food Regulations 2004,
The Food Safety Act 1990 (Amendment) Regulations 2004,
The Medicines Acts 1968 and 1971, 1981, 1986 & 2003,
The Food and Environmental Protection Act 1985,
The Consumer Protection Act 1987,
The Cosmetic (Safety) (Amendment) Regulations 2008,
Registration, Evaluation, Authorisation and Restriction of Chemicals (EC1970/2006),
-
Trade associations representing companies involved with such products.
-
Companies, partnerships or individuals who are directly concerned with research, development or marketing of a product which is being considered by the Committees on Toxicity, Mutagenicity, or Carcinogenicity of Chemicals in Food, Consumer Products and the Environment.
-
‘the Secretariat’ means the Secretariat of the COC, COM and COT.
-
‘the Agency’ means either the Food Standards Agency or the Health Protection Agency.
-
references to “member(s)” includes the Chair.
Different types of Interest
The following is intended as a guide to the kinds of interests which should be declared. Where members are uncertain as to whether an interest should be declared, they should seek guidance from the Secretariat or, where it may concern a particular product which is to be considered at a meeting, from the Chair at that meeting.
If members have interests not specified in these notes but which they believe could be regarded as influencing their advice they should declare them.
However, neither the members nor the Secretariat are under any obligation to search out links of which they might reasonably not be aware. This Code suggests that interests of close family members are declared, members have in the past limited such declarations to personal partners, parents, children (minor and adult), brothers, sisters and the personal partners of any of these with the emphasis on disclosure only where the interest may or may be perceived (by a reasonable member of the public) to influence a members’ judgement.
The Secretariat is required to publish an up-to-date register of members’ interests, and these can be found on the relevant Committees website.
Personal Interests
A personal interest involves the member personally. The main examples are:
-
Consultancies and/or direct employment: any consultancy, directorship, position in or work for industry which attracts regular or occasional payments in cash or kind.
-
Fee-Paid Work: any work commissioned by industry for which the member is paid in cash or kind.
-
Shareholdings: any shareholding in or other beneficial interest in shares of industry. This does not include shareholdings through unit trusts or similar arrangements where the member has no influence on financial management.
-
Membership or Affiliation: any membership role or affiliation that you or a close family member has to clubs or organisations with an interest or involvement in the work of the Agency.
Non-Personal Interests
A non-personal interest involves payment which benefits the organisation in which the member works but is not received by the member personally. The main examples are:
-
Fellowships: the holding of a fellowship endowed by industry.
-
Support by Industry: any payment, other support or sponsorship which does not convey any pecuniary or material benefit to a member personally, but which does benefit their position or organisation, e.g.
-
A grant for the running of a unit or department for which the member is responsible.
-
A grant or fellowship or other payment to sponsor a post or a member of staff or a post graduate research programme for which the member is responsible. This does not include financial assistance for students.
-
The commissioning of research or other work by, or advice from, staff who work in a unit for which the member is responsible.
Members are under no obligation to seek out knowledge of work done for, or on behalf of, the industry or other relevant bodies by departments in which they work, if they would not normally expect to be informed.
-
Trusteeships: where a member is a trustee of a charity with investments in industry, the Secretariat can agree with the member a general declaration to cover this interest rather than draw up a detailed portfolio.
At meetings members are required to declare relevant interests and to state whether they are personal or non-personal interests and whether they are specific or nonspecific to the matter, product or substance under consideration.
Specific Interests
A member must declare a personal specific interest if they have at any time worked on a matter, product or substance under consideration and have personally received payment for that work, in any form.
A member must declare a non-personal specific interest if they are aware that the organisation in which they work has at any time worked on the matter, product or substance under consideration, but they have not personally received payment for that work, in any form.
Non-specific Interests
A member must declare a personal non-specific interest if they have a current personal interest in a company concerned with a matter, product or substance under consideration, which does not relate specifically to the matter, product or substance under discussion.
A member must declare a non-personal non-specific interest if they are aware that the organisation in which they work is currently receiving payment from the company concerned which does not relate specifically to the matter, product or substance under discussion.
If a member is aware that a substance, product or matter under consideration is or may become a competitor of a substance, product or matter manufactured, sold or supplied by a company in which the member has a current personal interest, they should declare their interest in the company marketing the rival product, substance or matter.
Handling conflicts of interests
The purpose of these provisions is to avoid any danger of Committee members being influenced, or appearing to be influenced, by their private interests in the exercise of their public duties. All members should declare any personal or business interest which may or may be perceived (by a reasonable member of the public) to, influence their judgement. A guide to the types of interest that should be declared is mentioned above.
Declaration of Interests to the Secretariat
Members are required to inform the Agency in writing prior to appoint of their current personal and non-personal interests, including the principal position(s) held. Members are not required to disclose the amount of any salary, fee, shareholding, grant etc. An interest is current if the member has an on-going financial involvement e.g., if he or she holds shares in industry, has a consultancy contract, or if they or the organisation for which they are responsible is in the process of carrying out work for the industry.
Following appointment members are asked to inform the Secretariat at the time of any change in their personal interests. However, the Secretariat will contact each member on an annual basis to update their declaration of interests. Changes in non-personal interests can be reported annually, and those involving less than £1000 from a particular company in the previous year need not be declared. The register of interests is kept up-to-date and open to the public via the website.
Declaration of Interest at Meetings
Members of the Committee are required to verbally declare any direct interests relating to salaried employment or consultancies, or those of close family 8 members in matters under discussion at each meeting, and if items are taken by correspondence between meetings. The declaration should note whether the interest is personal or nonpersonal, whether it is specific to the item under discussion, or non-specific and whether it is current or lapsed. Having fully explained the nature of their interest the Chair will, decide whether and to what extent the member should participate in the discussion and determination of the issue, and it should be recorded in the minutes of the meeting.
Withdrawal from meetings
If a declaration of interest has been made and the Committee decides that the member should not participate in the discussion and should withdraw from the meeting (even if held in public) and it should be recorded in the minutes of the meeting. The Chair may first allow them to make a statement on the item under discussion.
Personal liability of Committee members
The Department of Health has a formal statement of indemnity for its advisory committee members, which includes the COC and COM, its guidance is taken from the Cabinet Office “Model Code of Practice for Board Members of Advisory Non-Departmental Public Bodies” and states that “Legal proceedings by a third party against individual board members of advisory bodies are very exceptional. A board member may be personally liable if he or she makes a fraudulent or negligent statement which result in a loss to a third party; or may commit a breach of confidence under common law or criminal offence under insider dealing legislation, if he or she misuses information gained through their position. However, the Government has indicated that individual board members who have acted honestly, reasonably, in good faith and without negligence will not have to meet out of their own personal resources any personal civil liability which is incurred in execution or purported execution of their board functions. Board members who need further advice should consult the sponsor department.”9 except where the person has acted recklessly.
The FSA has also drawn up a formal statement of indemnity for its advisory committee members.
Indemnity by the Food Standards Agency to Members of the
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment
The Food Standards Agency hereby undertakes with the members (including the Chair) of the Committee on Toxicity of Chemicals in food, Consumer products and the environment (COT) to indemnify them against all liability in respect of any action or claim which may be brought, or threatened to be brought, against them either individually or collectively by reason of or in connection with the performance of their duties as members, including all costs, charges and expenses which the Members may properly and reasonably suffer or incur in disputing any such action or claim.
The Members shall as soon as practicable notify the Food Standards Agency if any action or claim is brought or threatened to be brought against them in respect of which indemnity may be sought and if an action or claim is brought, the Food Standards Agency shall be entitled to take conduct of the defence, dispute, compromise or appeal of the action or claim and of any incidental negotiations relating to the action or claim.
The Food Standards Agency shall notify the Members as soon as practicable if it intends to so take conduct and the Members shall then provide to the Food Standards Agency such information and assistance as it shall reasonably request, subject to all out of pocket expenses properly and reasonably incurred by them being reasonably reimbursed. The Food Standards Agency shall, to the extent reasonable and practicable, consult with and keep the Members informed as and when reasonably requested by the Members in respect of any action or claim. If the Food Standards Agency does not so take conduct the Members shall keep the Food Standards Agency fully informed of the progress of the action or claim and any consequent legal proceedings and consult with the Food Standards Agency as and when required by the Food Standards Agency concerning the action or claim.
The indemnity shall not extend to any losses, claims, damages, costs, charges, expenses and any other liabilities:
- In respect of which the Members are indemnified by or through any defence organisation or insurers or,
- which may result from bad faith (including dishonesty), wilful default or recklessness on the part of the Members,
- which may result from any of the following circumstances:
- any settlement made or compromise effected without the knowledge or consent of the Food Standards Agency on behalf of the Members of any action or claim brought, or threatened to be brought, against the Members.
- Any admission by the Members of any liability or responsibility in respect of any action or claim brought, or threatened to be brought, against them.
- Members taking action that they were aware, or ought reasonably to have been aware, might prejudice the successful defence of any action or claim, once the Members had become aware that such an action or claim had been brought or was likely to be brought.
Remuneration and Committee finance
In the financial year 2020/2021 the budget for the COT, excluding Secretariat resources was £87,800. Costs were met by the Food Standards Agency (FSA).
Committee members may claim a fee for Committee meetings:
COT Committee Chair £400 per day
COT Committee Member £300 per day
COT Members are able to claim for work undertaken between meetings at the above rates.
Different provisions apply to COC and COM Members. Details can be obtained from their respective Secretariats.
Review of fee rates
Fees in respect of the COT are set by the FSA and for COC and COM by the Department of Health and Social Care. The FSA will review and revise COT rates every 2 years with the intention that rates should rise in line with the recommendations of the Senior Salaries Review Board with regard to pay in the Senior Civil Service. The FSA will also take into account comparisons with rates paid in similar advisory bodies in the UK.
Travel and other expenses
Committee members are entitled to reimbursement of reasonable travel and subsistence expenses necessarily incurred on official committee business. Members must seek value for money and are encouraged to use the most cost effective and environmentally sustainable options for travel and accommodation.
Working Groups
The Committee may establish Working Groups to consider particular topics in depth or to make brief assessments of particular issues and advise the main Committee on the possible need for further action. Such Groups contain a number of Committee members (supplemented, as necessary, by external expertise in the particular subject being considered). A Committee Chair will play a leading role in deciding which Committee members should be invited to join such groups, which may meet on a number of occasions in a particular year. Committee members may claim an allowance for participating on a Working Group.
Terms and conditions of appointment
Appointments of members may be staggered so that only a proportion retire or are re-appointed each year, to help ensure continuity. COC and COM Chairs are ex officio members of each other’s Committees.)
COC and COM members are usually expected to attend 3 meetings in a year. COT members are expected to attend 7 meetings in a year. Members should allow appropriate preparation time. Meetings will usually be in London.
The COC/COM/COT Chair must also be available for a number of other activities including: attending, with the FSA Chief Scientist, the FSA Board’s annual discussion of the Agency’s science; engaging with the media on any high-profile relating to the Committee’s work, and discussion with the Agency Chief Scientist and GACS Secretariat in planning and developing the Committee’s work (including discussing and agreeing with the Agency’s Chief Scientist a framework for providing assurance on the work of the Scientific Advisory Committees in providing advice to the Agency). It is expected that these additional activities might require 5-10 days input per year.
Feedback on performance
The COT Chair and members are asked to provide brief feedback on their experience on the committee each year to help the Agency ensure that the Committee operates effectively and identify any areas for improvement.
Committee members are normally appointed for a term of 3 years (a maximum 10 years/3 terms per member). The COT uses the feedback self-assessment form as one of the tools used to determine whether or not a committee member should be reappointed at the end of their (3 year) term.
Annex 3 – Openness
In this guide
In this guideIntroduction
1. The Committee on Toxicity (COT) and its sister committees the Committee on Mutagenicity (COM) and Committee on Carcinogenicity (COC) are non-statutory independent scientific advisory committees which advise the Chair of the Food Standards Agency and the Chief Medical Officers (for England, Scotland, Wales and Northern Ireland) and, through them, the Government on a wide range of matters concerning chemicals in food, consumer products and the environment.
2. The Government is committed to make the operation of scientific advisory committees such as the COT/COM/COC hereafter referred to as “the Committee” more open and to increase accountability. The Committee is aware that the disclosure of information that is of a confidential nature and is communicated in circumstances importing an obligation of confidence is subject to the common law of confidentiality. There are some circumstances making disclosure of confidential information lawful for example, where the individual to whom the information relates has consented; where disclosure is in the public interest; and where there is a legal duty to do so. However, guidance is set out in the Freedom of Information Act 2000 which gives any person legal rights of access to information which is held by a public authority.
3. The Committee has agreed to hold open meetings as standard practice. Interest groups, consumer organisations etc can attend (subject to the appropriate procedures for handling commercially sensitive information and research not in the public domain, paragraphs 9-15 refer).
4. The Committee appoints lay/public interest member(s) to help to increase public scrutiny of Committee business.
5. The Committee has agreed to the publication of agendas, draft and finalised minutes, discussion papers and statements on the internet.
6. Statements will summarise all the relevant data, such as information regarding potential hazards/risks for human health in respect of the use of products and chemicals, and any recommendations for further research.
7. The Committee will be asked for an opinion based on the data available at the time of consideration. It is recognised that, for many chemicals, the toxicological information is incomplete and that recommendations for further research to address these gaps may form part of the Committee's advice.
8. The release of documents (papers, minutes and statements) where the Committee has agreed an opinion on the available unpublished data but where further additional information is required in order to finalise the Committee's conclusions, needs to be considered on a case-by case basis.
The relevant considerations include the likelihood that such additional data would alter the Committee's conclusion, any representations made by a company about, for example, commercial harm that early disclosure could cause and also the public interest in disclosure.
Procedures for handling commercially sensitive information and research data not in the public domain
Background
9. The Committee operates on a presumption of openness. However, it is recognised that the nature of the work will at times provide the Committee access to information that is not in the public domain. Decisions on confidentiality will be exercised consistently with consideration to the Freedom of Information Act 2000 and Environmental Information Regulations 2004.
10. Where there is a need to discuss matters that cannot be put in the public domain the Committee may hold a discussion in “Reserved Business”. These items will be generally discussed either at the beginning or the end of an open meeting. It is expected that such cases will be infrequent and only in clearly justified circumstances. For the most part this comprises information which is commercially sensitive such as product formulations/specifications, methods of manufacture, and reports of toxicological investigations and company evaluations and safety assessment. It would also include pre-publication or unpublished research data.
11. “Reserved Business” items will be clearly indicated as such. The Committee will advise its reasons for withholding any information, and, if possible, an indication of when and where the information withheld may be published. Information subject to such restriction, including reserved sections of the minutes will be placed in the public domain as soon as practicable should the restrictions cease to apply at a later date.
12. Normal procedure is to publish a summary of the Committee's advice on their respective websites, in the Annual Report and where necessary to ask companies to release full copies of submitted reports for retention by the British Library at the completion of a review. Given the clear Ministerial commitment to the publication of detailed information regarding the activities of advisory committees, and in particular following the assessment of products which are already available to the general public, the Committee will publish statements via the Internet soon after they have been finalised.
13. Except in cases where there is legislation under which information has been submitted and which deals with disclosure and non-disclosure, the general principle of the common law duty of confidentiality will apply. This means that any information which is commercially sensitive, pre-publication or unpublished research data and has been obtained in circumstances importing a duty of confidence may not be disclosed unless consent has been given or there is an overriding public interest in disclosure (such as the prevention of harm to others).
14. The following procedure will be adopted which allows commercially sensitive information to be identified, assessed and appropriate statements to be drafted and published on the basis of a prior mutual understanding with the companies. There is scope for companies to make representations also after submission of the information and prior to publication regarding the commercial sensitivity of data supplied and to comment on the text of statements which are to be published. However, companies would not have a right of veto in respect of such statements.
Procedures prior to committee consideration
Initial discussions
15. Upon referral to Committee the Secretariat will liaise with the relevant company supplying the product in the UK to:
-
Clearly state the policy of Committee openness (summarised above).
-
Identify and request the information needed by the Committee (e.g., test reports, publications etc).
Commercially sensitive information
16. The company will be asked to clearly identify any commercially sensitive information and the reason for confidentiality.
Pre-publication and unpublished research data
17. The Committee and Secretariat will respect the confidentiality of authors of (unpublished or pre-publication) research data.
Handling confidential data
18. The procedures by which the Committee will handle commercially sensitive information, pre-publication or unpublished research data and the public availability of papers, minutes, conclusions and statements where reference is made to such data will be discussed with the company or author prior to submission of papers to the Committee and is outlined in paragraphs 9-15 above. Companies will be informed that confidential annexes to Committee papers (e.g. where detailed information supplied in confidence such as individual patient information and full study reports of toxicological studies) will not be disclosed but that other information will be disclosed unless agreed otherwise with an individual company.
19. The following is a suggested list of information which may be disclosed in Committee documents (papers, minutes and statements). The list is not exhaustive and is presented as a guide:
- name of product (or substance/chemical under consideration),
- information on physico-chemical properties,
- methods of rendering harmless,
- a summary of the results and evaluation of the results of tests to establish harmlessness to humans,
- methods of analysis,
- first aid and medical treatment to be given in the case of injury to persons,
- surveillance data (e.g. monitoring for levels in food, air, or water).
Procedures during and after Committee consideration
20. The timing of release of Committee documents (papers, minutes and statements) where the item of business involved the consideration of confidential data would be subject to the general provisions outlined in paragraphs 9-15 above. Documents would not be released until the Committee statement is available.
21. The most important outcome of the Committee consideration is likely to be the agreed statement. Companies will be given an opportunity to comment on the statement prior to publication and to make representations (for example, as to commercial sensitivities in the statement). The Chair would be asked to consider any comments provided, but companies would not be able to veto the publication of a statement or any part of it. Companies will continue to be asked to release full copies of submitted reports for retention by the British Library at the completion of a review.
Dissenting views
22. The Committee should not seek consensus at the risk of failing to recognise different views on a subject. Any significant diversity of opinion among the members of the Committee that cannot be resolved should be accurately reflected in the minutes or report. Committee decisions should always include an explanation of where differences of opinion have arisen during discussions, specifically where there are unresolved issues and why conclusions have been reached. If however member(s) feel they cannot support the Committee conclusions they may declare a ‘minority report’ identifying which member(s) are making the minority report and setting out their position.
COC/COM/COT papers
23. Committee papers are available on the respective website. Papers will not include commercially sensitive documents, pre-publication, unpublished or material in the public domain. Where possible a cover page with weblinks (current at the time) will be provided.
Annex 4 – Good Practice Agreement for Scientific Advisory Committees
In this guide
In this guideIntroduction
The Government Chief Scientific Adviser’s Guidelines on the Use of Scientific and Engineering Advice in Policy Making set out the basic principles which government departments should follow in assembling and using scientific advice. The key elements are to:
- Identify early the issues which need scientific and engineering advice and where public engagement is appropriate.
- Draw on a wide range of expert advice sources, particularly when there is uncertainty.
- Adopt an open and transparent approach to the scientific advisory process and publish the evidence and analysis as soon as possible.
- Explain publicly the reasons for policy decisions, particularly when the decision appears to be inconsistent with scientific advice.
- Work collectively to ensure a joined-up approach throughout government to integrating scientific and engineering evidence and advice into policy making.
The Code of Practice for Scientific Advisory Committees and the Principles of Scientific Advice to Government provide more detailed guidance on the operation of Scientific Advisory Committees (SACs) and their relationship with their sponsor Departments.
The Food Standards Agency’s Board adopted a Science Checklist in 2006 (updated in 2012) that makes explicit the points to be considered in the preparation of policy papers and proposals dealing with science-based issues, including those which draw on advice from the SACs.
These Good Practice Guidelines were drawn up in 2006 by the Chairs of the independent SACs that advise the FSA based on, and complementing, the Science Checklist. They were updated in 2012 in consultation with the General Advisory Committee on Science (GACS). (Note GACS has now been replaced by the FSA Science Council).
The Guidelines apply to the SACs that advise the FSA and for which the FSA is sole sponsor Department:
- Advisory Committee on Animal Feedingstuffs,
- Advisory Committee on Microbiological Safety of Food,
- Advisory Committee on Novel Foods and Processe,
- Science Council,
- Advisory Committee for Social Science (ACSS).
As well as those Committees, the FSA co-sponsors with the Department of Health and Social Care:
- Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment,
- Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment,
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment.
For the SACs with a shared sponsorship the Guidelines apply formally to their advice to the FSA; they may opt to follow them also in advising other sponsor Departments.
All these committees share important characteristics. They are:
- Independent,
- work in an open and transparent way,
- are concerned with risk assessment and/or science governance, not with decisions about risk management.
The Guidelines relate primarily to the risk assessment process since this is the main purpose of most of the SACs. However, the SACs may, where appropriate, comment on risks associated with different risk management options, highlight any wider issues raised by their assessment that they feel should be considered (distinguishing clearly between issues on which the SAC has an expert capability and remit, and any other issues), or any evidence gaps and/or needs for research or analysis.
In addition, the Science Council and ACSS may advise the FSA on aspects of the governance of risk management, or on research that relates to risk management.
Twenty-nine principles of good practice have been developed. However, the different committees have different duties and discharge those duties in different ways. Therefore, not all the principles set out below will be applicable to all of the committees, all of the time.
The SACs have agreed to review their application of the principles annually and report this in their Annual Reports. Compliance with the Guidelines will also be covered in the annual self-assessments by Members and annual feedback meetings between each SAC Chair and the FSA Chief Scientist.
Principles
Defining the problem and the approach
The FSA will ensure that issues it asks an SAC to address are clearly defined and take account of stakeholder expectations in discussion with the SAC Secretariat and where necessary the SAC Chair. The SAC Chair will refer back to the FSA if discussion suggests that further iteration and discussion of the task is necessary.
Where an SAC proposes to initiate a piece of work the SAC Chair and Secretariat will discuss this with FSA to ensure the definition and rationale for the work and its expected use by the FSA are clear.
Seeking input
The Secretariat will ensure that stakeholders are consulted at appropriate points in the SAC’s considerations. It will consider with the FSA whether and how stakeholder views need to be taken into account in helping to identify the issue and frame the question for the committee.
Wherever possible, SAC discussions should be held in public.
The scope of literature searches made on behalf of the SAC will be clearly set out.
Steps will be taken to ensure that all available and relevant scientific evidence is rigorously considered by the committee, including consulting external/additional scientific experts who may know of relevant unpublished or pre-publication data.
Data from stakeholders will be considered and weighted according to quality by the SAC.
Consideration by the Secretariat and the Chair (and where appropriate the whole SAC) will be given to whether expertise in other disciplines will be needed.
Consideration will be given by the Secretariat or by the SAC, in discussion with the FSA, as to whether other SACs need to be consulted.
Validation
Study design, methods of measurement and the way that analysis of data has been carried out will be assessed by the SAC.
Data will be assessed by the committee in accordance with the relevant principles of good practice, e.g. qualitative social science data will be assessed with reference to guidance from the Government’s Chief Social Researcher as set out in Quality in qualitative Evaluation: A Framework for Assessing Research Evidence or the Magenta Book
Formal statistical analyses will be included wherever appropriate. To support this, each SAC will have access to advice on quantitative analysis and modelling as needed.
When considering what evidence needs to be collected for assessment, the following points will be considered:
- the potential for the need for different data for different parts of the UK or the relevance to the UK situation for any data originating outside the UK.
- whether stakeholders can provide unpublished data.
The list of references will make it clear which references have been subject to external peer review, and which have been peer reviewed through evaluation by the Committee, and if relevant, any that have not been peer reviewed.
Uncertainty
When reporting outcomes, SACs will make explicit the level and type of uncertainty (both limitations on the quality of the available data and lack of knowledge) associated with their advice.
Any assumptions made by the SAC will be clearly spelled out, and, in reviews, previous assumptions will be challenged.
Data gaps will be identified and their impact on uncertainty assessed by the SAC.
An indication will be given by the SAC about whether the evidence base is changing or static, and if appropriate, how developments in the evidence base might affect key assumptions and conclusions.
Drawing conclusions
The SAC will be broad-minded, acknowledging where conflicting views exist and considering whether alternative interpretations fit the same evidence.
Where both risks and benefits have been considered, the committee will address each with the same rigour, as far as possible; it will make clear the degree of rigour and uncertainty, and any important constraints, in reporting its conclusions.
SAC decisions will include an explanation of where differences of opinion have arisen during discussions, specifically where there are unresolved issues, and why conclusions have been reached. If it is not possible to reach a consensus, a minority report may be appended to the main report, setting out the differences in interpretation and conclusions, and the reasons for these, and the names of those supporting the minority report.
The SAC’s interpretation of results, recommended actions or advice will be consistent with the quantitative and/or qualitative evidence and the degree of uncertainty associated with it.
SACs will make recommendations about general issues that may have relevance for other committees.
Communicating the SACs’ conclusions
Conclusions will be expressed by the SAC in clear, simple terms and use the minimum caveats consistent with accuracy.
It will be made clear by the SAC where assessments have been based on the work of other bodies and where the SAC has started afresh and there will be a clear statement of how the current conclusions compare with previous assessments.
The conclusions will be supported by a statement about their robustness and the extent to which judgement has had to be used.
As standard practice, the SAC secretariat will publish a full set of references (including the data used as the basis for risk assessment and other SAC opinions) at as early a stage as possible to support openness and transparency of decision-making. Where this is not possible, reasons will be clearly set out, explained and a commitment made to future publication wherever possible.
The amount of material withheld by the SAC or FSA as being confidential will be kept to a minimum. Where it is not possible to release material, the reasons will be clearly set out, explained and a commitment made to future publication wherever possible.
Where proposals or papers being considered by the FSA Board rest on scientific evidence produced by a SAC, the Chair of the SAC (or a nominated expert member) will be invited to the table at the Open Board meetings at which the paper is discussed. To maintain appropriate separation of risk assessment and risk management processes, the role of the Chairs will be limited to providing an independent view and assurance on how their committee’s advice has been reflected in the relevant policy proposals, and to answer Board Members’ questions on the science. The Chairs may also, where appropriate, be invited to provide factual briefing to Board members about particular issues within their committees’ remits, in advance of discussion at open Board meetings.
The SAC will seek (and FSA will provide) timely feedback on actions taken (or not taken) in response to the SAC’s advice, and the rationale for these.
Annex 5 – Glossary of Terms
In this guide
In this guide3R’s principle:
The 3Rs stand for Replacement, Reduction, Refinement. This is a strategy that is intended to reduce the number of animals used in experiments and to reduce animal experimentation overall; it also aims to mitigate the suffering and distress caused to the animals.
a priori:
The formulation of an hypothesis based on theoretical considerations before undertaking an investigation or experiment.
Absolute risk (AR):
is the probability or chance of an event. It is usually used for the number of events (such as a disease) that occurred in a group, divided by the number of people in that group.
Absorption (biological):
Process of active or passive transport of a substance into an organism, in humans this is usually through the lungs, gastrointestinal tract or skin.
Acceptable daily intake (ADI):
Estimate of the amount of a substance in food or drink, expressed on a bodyweight basis (e.g. mg/kg bodyweight), that can be ingested daily over a lifetime by humans without appreciable health risk.
Acceptable risk:
Probability of suffering disease or injury which is considered to be sufficiently small to be societally acceptable.
Acute:
Short term, in relation to exposure or effect.
Acute reference dose (ARfD):
Estimate of the amount of a substance in food or drink, expressed on a body weight basis that can be ingested in a period of 24 hours or less without appreciable health risk.
Acute toxicity:
Adverse effects that occur over a short period of time (up to 14 days) immediately following a single exposure.
Adaptive response:
The process whereby a cell or organism responds to a xenobiotic so that the cell or organism will survive in the new environment that contains the xenobiotic without impairment of function.
Adduct:
A chemical grouping which is covalently bound (see covalent binding) to a large molecule such as DNA (qv) or protein.
Adductome:
The totality of the adduct profile, usually to DNA, in an individual.
Adenoma:
A benign neoplasm arising from a gland forming epithelial tissue such as colon, stomach or respiratory tract.
Adverse Outcome Pathway (AOP):
A sequence of key events linking a molecular initiating event (MIE) to an adverse outcome through different levels of biological organisation. AOPs span multiple layers of biological organisation.
Adverse response:
Change in morphology, physiology, biochemistry, growth, development or lifespan of an organism or its progeny which results in impairment of functional capacity or impairment of capacity to compensate for additional stress or increase in susceptibility to the harmful effects of other environmental influences.
Aetiology:
study of causation or origination.
Aggregate exposure:
exposure to one chemical by all routes from all sources.
Ah receptor:
The Ah (Aromatic hydrocarbon) receptor protein is a member of a group of regulatory sensor molecules. The identity of the natural endogenous chemicals which regulate the Ah receptor is unknown. Binding to the Ah receptor is an integral part of the toxicological mechanism of a range of chemicals, such as chlorinated dibenzodioxins and polychlorinated biphenyls.
Alkylating agents:
Chemicals which leave an alkyl group covalently bound to biologically important molecules such as proteins and nucleic acids (see adduct). Many alkylating agents are mutagenic, carcinogenic and immunosuppressive.
Allele:
Alternative form of a gene within the population.
Allergen:
Substance capable of stimulating an allergic reaction.
Allergy:
The adverse health effects that may result from the stimulation of a specific immune response.
Allergic reaction: an adverse reaction elicited by exposure to a previously sensitised individual to the relevant antigen.
Ames test:
Also known as the bacterial reverse mutation assay. In vitro assay for bacterial gene mutations using strains of Salmonella typhimurium and Escherichia coli.
Androgen:
The generic term for any natural or synthetic compound that can interact with and activate the androgen receptor. In mammals, androgens (for example, androstenedione and testosterone) are synthesised by the adrenal glands and the testes and promote development and maintenance of male secondary sexual characteristics.
Aneugen/aneugenic:
(An agent) Inducing aneuploidy.
Aneuploidy:
The circumstances in which the total number of chromosomes within a cell is not an exact multiple of the normal haploid (see 'polyploidy') number. Chromosomes may be lost or gained during cell division.
Apoptosis:
A form of programmed, active cell death resulting in fragmentation of the cell into membrane-bound fragments (apoptotic bodies). These are usually rapidly removed in vivo by engulfment by phagocytic cells. Apoptosis occurs normally during development but can be triggered abnormally by toxic stimuli.
As low as is reasonably achievable/ As low as is reasonably practicable (ALARA/ALARP):
A risk management approach under which exposure to a substance or mixture is reduced to the lowest level that it is deemed to be reasonably achievable or practicable in particular circumstances or by available technological solutions.
Base pair (bp):
Two complementary nucleotide bases in DNA joined together by hydrogen bonds.
Benchmark dose (BMD) modelling:
An alternative quantitative approach to dose-response assessment using more of the data than the NOAEL process. This approach utilises mathematical models to fit all available data points and uses the best fitting model to interpolate an estimate of the dose (benchmark dose) that corresponds to a particular level of response (a benchmark response). A measure of uncertainty is also calculated, and the lower confidence limit on the benchmark dose is called the BMDL. The BMDL accounts for the uncertainty in the estimate of the dose-response that is due to characteristics of the experimental design such as sample size and biological variability. The BMDL can be used as the point of departure (see POD) for derivation of a health-based guidance value or a margin of exposure.
Benign tumour:
Tumours showing a close morphological resemblance to their tissue of origin, growing in a slow expansile fashion and with a circumscribed form, usually encapsulated masses. They may stop growing and they may regress. Benign tumours do not infiltrate through local tissues, and they do not metastasise. They are rarely fatal.
Bias:
An interference which at any stage of an investigation tends to produce results that depart systematically from the true values (to be distinguished from random error). The term does not necessarily carry an imputation of prejudice or any other subjective factor such as the experimenter's desire for a particular outcome.
Bioavailability:
A term referring to the proportion of a substance which reaches the systemic circulation unchanged after a particular route of administration.
Bioinformatics:
The science of informatics as applied to biological research. Informatics is the management and analysis of data using advanced computing techniques. Bioinformatics is particularly important as an adjunct to -omics research, because of the large amount of complex data this research generates.
Biological relevance:
an effect considered by expert judgement as important and meaningful for human, animal, plant or environmental health. It therefore implies a change that may alter how decisions for a specific problem are taken.
Biomarker:
Observable change (not necessarily pathological) in an organism, related to a specific exposure, effect or susceptibility.
Biomarker of effect:
A measurable biochemical, physiologic, behavioural, or other alteration in an organism that, depending on the magnitude, can be recognised as associated with an established or possible health impairment or disease.
Biomarker of exposure:
a chemical, its metabolite, or the product of an interaction between a chemical and some target molecule or cell that is measured in the human body indicative of exposure.
Biomarker of susceptibility:
An indicator of an inherent or acquired ability of an organism to respond to the challenge of exposure to a specific chemical substance.
Biomonitoring (human):
the direct measurement of people's integrated exposure to toxic substances by measuring the substances, their metabolites or a biochemical change in human specimens, such as blood or urine.
Biomonitoring equivalent:
an estimated concentration or range of concentrations of an environmental chemical in humans which is consistent with existing health-based guidance values such as the TDI or RfD/RfC. BEs provide a way of interpreting biomonitoring data in the context of these values.
Body burden:
Total amount of a chemical present in an organism at a given time.
Bradford Hill considerations:
Sir Austin Bradford Hill established a set of ‘principles’ (not be taken as ‘criteria’) that may be used to assist in the interpretation of associations reported from epidemiological studies:
Strength – The stronger the association the more likely it is causal. The COC has previously noted that the relative risks of <3 need careful assessment for effects of bias or confounding.
Consistency – The association has been consistently identified by studies using different approaches and is also seen in different populations with exposure to the chemical under consideration.
Specificity – Limitation of the association to specific exposure groups or to specific types of disease increases likelihood that the association is causal.
Temporality – The association must demonstrate that exposure leads to disease. The relationship of time since first exposure, duration of exposure and time since last exposure are all important in assessing causality.
Biological gradient – If an association reveals a biological gradient or dose response curve, then this evidence is of particular importance in assessing causality.
Plausibility – Is there appropriate data to suggest a mechanism by which exposure could lead to concern? However, even if an observed association may be new to science or medicine it should not be dismissed.
Coherence – Cause and effect interpretation of data should not seriously conflict with generally known facts.
Experiment – Can the association be demonstrated experimentally? Evidence from experimental animals may assist in some cases. Evidence that removal of the exposure leads to a decrease in risk may be relevant.
Analogy – Have other closely related chemicals been associated with the disease?
Bronchial:
Relating to the air passages conducting air from the trachea (windpipe) to the lungs.
Cancer:
Synonym for a malignant neoplasm – that is, a tumour that grows progressively, invades local tissues and spreads to distant sites (see also tumour and metastasis).
Candidate gene:
A gene that has been implicated in causing or contributing to the development of a particular disease.
Carcinogen:
A causal agent that induces tumours. Carcinogens include external factors (chemicals, physical agents, viruses) and internal factors such as hormones. An important distinction can be drawn between genotoxic carcinogens which have been shown to damage DNA, and nongenotoxic carcinogens which act through other mechanisms. The activity of genotoxic carcinogens can often be predicted from their chemical structure - either of the parent compound or of active metabolites. Most chemical carcinogens exert their effects after prolonged exposure, show a dose-response relationship and tend to act on a limited range of susceptible target tissues. Carcinogens are sometimes species or sex-specific and the term should be qualified by the appropriate descriptive adjectives to aid clarity. Several different chemical and other carcinogens may interact, and constitutional factors (genetic susceptibility, hormonal status) may also contribute, emphasising the multifactorial nature of the carcinogenic process.
Carcinoma:
Malignant tumour arising from epithelial cells lining, for example, the alimentary, respiratory and urogenital tracts and from epidermis, also from solid viscera such as the liver, pancreas, kidneys and some endocrine glands. (See also 'tumour').
Case-control study:
(Synonyms - case comparison study, case referent study) A comparison is made of the proportion of cases who have been exposed to a particular hazard (e.g., a carcinogen) with the proportion of controls who have been exposed to the hazard.
Cell cycle (cell cycle arrest):
The cell cycle is a series of events involving the growth, replication, and division of a eukaryotic cell. Cell cycle arrest: A regulatory process that halts progression through the cell cycle during one of the normal phases (G1, S, G2, M).
Cell transformation:
The process by which a normal cell acquires the capacity for neoplastic growth. Complete transformation occurs in several stages both in vitro and in vivo. One step which has been identified in vitro is 'immortalisation' by which a cell acquires the ability to divide indefinitely in culture. Such cells do not have the capacity to form tumours in animals but can be induced to do so by extended passage in vitro, by treatment with chemicals, or by transfection with oncogene DNA. The transformed phenotype so generated is usually, but not always, associated with the ability of the cells to grow in soft agar and to form tumours when transplanted into animals. It should be noted that each of these stages of transformation can involve multiple events which may or may not be genetic. The order in which these events take place, if they occur at all, in vivo is not known.
Cholinergic:
A substance which is capable of producing, altering or releasing the neurotransmitter acetylcholine.
Chromosomal aberrations:
Collective term of particular types of chromosome damage induced after exposure to exogenous chemical or physical agents which damage the DNA (see also aneugen, clastogen). Such numerical or structural chromosome changes tend to be those which are evident using light microscopy.
Chromosome:
In simple prokaryotic organisms, such as bacteria and most viruses, the chromosome consists of a single circular molecule of DNA containing the entire genetic material of the cell. In eukaryotic cells, the chromosomes are thread-like structures, composed mainly of DNA and protein, which are present within the nuclei of every cell. They occur in pairs, the numbers varying from one to more than 100 per nucleus in different species. Normal somatic cells in humans have 23 pairs of chromosomes, each consisting of linear sequences of DNA which are known as genes.
Chronic effect:
Consequence which develops slowly and has a long-lasting course (often but not always irreversible).
Chronic exposure:
Continued exposures occurring over an extended period of time, or a significant fraction of the life-time of a human or test animal.
Clastogen:
An agent that produces chromosome breaks and other structural aberrations such as translocations. Clastogens may be viruses or physical agents as well as chemicals. Clastogenic events play an important part in the development of some tumours (clastogenicity).
Clearance:
Volume of blood or plasma, or mass of an organ, effectively cleared of a substance by elimination (metabolism and excretion) in a given time interval. Total clearance is the sum of the clearances for each eliminating organ or tissue.
Clone:
A term which is applied to genes, cells, or entire organisms which are derived from - and are genetically identical to - a single common ancestor gene, cell, or organism, respectively. Cloning of genes and cells to create many copies in the laboratory is a common procedure essential for biomedical research.
Coding regions:
those parts of the DNA that contain the information needed to form proteins. Other parts of the DNA may have non-coding functions (e.g., start-stop, pointing or timer functions) or as yet unresolved functions or maybe even ‘noise’.
Codon:
a set of three nucleotide bases in a DNA or RNA sequence, which together code for a specific amino acid.
Cohort:
A defined population that continues to exist through a period of time, e.g., a group of individuals who had a specific occupation.
Cohort study:
(Synonyms - follow-up, longitudinal study) The study of a group of people defined at a particular point in time (the cohort), who have particular characteristics in common, such as a particular exposure. They are then observed over a period of time for the occurrence of disease. The rate at which the disease develops in the cohort is compared with the rate in a comparison population, in which the characteristics (e.g., exposure) are absent.
Combined exposure:
exposure to multiple chemicals by a single or multiple routes at the same or different times.
Comet assay:
A genotoxicity assay in which DNA strand breaks in an individual cell are measured using single-cell gel electrophoresis. Cell DNA fragments assume a "comet with tail" formation on electrophoresis and are detected with an image analysis system. Alkaline assay conditions facilitate sensitive detection of double-strand and single-strand damage, as well as alkali-labile sites. Modifications to standard methodology enable detection of types of DNA damage, e.g., DNA-DNA or DNA-protein cross-links and base-oxidation.
Complementary DNA (cDNA):
cDNA is DNA that is synthesised in the laboratory from mRNA by reverse transcription. A cDNA is so-called because its sequence is the complement of the original mRNA sequence.
Confounding variable:
(synonym - confounder) A confounding variable is a factor that is independently associated with both an intervention or exposure and the outcome of interest. Failure to account for this will distort the observed measure of association in the statistical analysis. For example, in observational studies, cigarette smoking is a confounding variable with respect to an association between alcohol consumption and heart disease because it is a risk factor for heart disease and is associated with alcohol consumption but is not a consequence of alcohol consumption. Similarly, if people in the experimental group of a controlled trial are younger than those in the control group, age could act as a potential confounder and make it difficult to ascertain whether a lower risk of death in one group is due to the intervention or the difference in ages.
Confounding may also occur in experimental studies, where in a feed trial, unpalatability might result in reduced food consumption and weight loss, rather than weight loss occurring through toxicity.
Congeners:
Related compounds varying in chemical structure that often, but not always, share biological properties.
Continuous Data:
Quantitative data that can be measured and has an infinite number of possible values within a selected range.
Copy number variants (CNVs):
Alterations in the DNA of a genome that results in the cell having an abnormal number of copies of one or more sections of the DNA. CNVs correspond to relatively large regions of the genome that have been deleted (fewer than the normal number) or duplicated (more than the normal number) on certain chromosomes.
Covalent binding:
Chemical bonding formed by the sharing of an electron pair between two atoms. Molecules are combinations of atoms bound together by covalent bonds (see adduct).
Critical effect size (CES):
The magnitude of the adverse effect selected at which to determine the dose to serve as a point of departure in assessing the risk from exposure to a chemical. This term is often used synonymously with Benchmark Response (BMR). Choice of CES includes both statistical and toxicological considerations.
Cumulative exposure:
exposure to multiple chemicals on the basis of grouping them on some common characteristic, such as mode of action, adverse effect, or inclusion in a product formulation.
P450 (CYP):
An extensive family of haem-containing proteins involved in enzymic oxidation of a wide range of endogenous and xenobiotic substances and their conversion to forms that may be more easily excreted. In some cases, the metabolites produced may be chemically reactive and have increased toxicity. In other cases, the substances may be natural precursors of hormones (e.g., steroids).
Cytogenetic:
Concerning chromosomes, their origin, structure and function.
(DNA) Deletion:
A type of mutation where there is a loss of DNA (nucleotide base pairs) from the genome. Deletions may range in size from a single nucleotide to an entire chromosome. Such deletions may be harmless, may result in disease, or may in rare cases be beneficial.
Deoxyribonucleic acid (DNA):
The carrier of genetic information for all living organisms except the group of RNA viruses. Each of the 46 chromosomes in normal human somatic cells consists of 2 strands of DNA containing an estimated 50 - 250 million nucleotides, specific sequences of which make up genes. DNA itself is composed of two interwound chains of linked nucleotides.
DNA damage:
Injuries to DNA that introduce deviations from its normal, chemical structure and which may, if left unrepaired, result in a mutation or a block of DNA replication. These deviations can occur naturally or may be caused by environmental physical or chemical agents.
DNA methylation:
A reversible biochemical modification of DNA more or less universally present in organisms from bacteria to humans. Methyl groups can be enzymatically added to or removed from cytosine (C). It is associated with silencing of DNA sequences.
DNA probe:
A piece of single-stranded DNA, typically labelled so that it can be detected (for example, a radioactive or fluorescent label can be used), which can single out and bind with another specific piece of DNA. DNA probes can be used to determine which sequences are present in a given length of DNA or which genes are present in a sample of DNA.
DNA repair:
Processes that repair potentially damaging changes in DNA, including those induced by chemical mutagens (see mutagen.) Through the action of enzymes, individual DNA bases may be replaced, or part of a strand of DNA may be replaced, using its opposite, paired strand as a template. These processes may themselves be prone to error and result in potentially deleterious changes.
DNA repair genes:
Genes which code for proteins that repair damage in DNA sequences.
DNA damage response (DDR):
Cells respond to the perception of DNA damage by arresting cell-cycle progression and attempting repair: collectively these actions are known as the DNA-damage response (DDR).
DNA sequencing:
process by which the sequence of nucleotides along a strand of DNA is determined. Where either the whole genome or the exome (the region which encodes proteins) is sequenced this is referred to as whole genome/exome sequencing (WGS/WES).
Dominant lethal mutation:
A dominant mutation (i.e., where mutation of a single allele is sufficient to cause a change in phenotype) that causes death of an early embryo.
Dopaminergic:
Releasing or involving dopamine as a neurotransmitter.
Dose:
Total amount of a substance administered to, taken or absorbed by an organism. May be qualified such as external dose, absorbed dose.
Dose-response relationship:
how an effect caused by a chemical changes as the dose of the chemical changes, after a certain exposure time.
Endocrine active substance (EAS):
A substance that can interact or interfere with the endocrine system.
Endocrine disrupter (ED):
An exogenous substance or mixture that alters function(s) of the endocrine system and consequently causes adverse health effects in an intact organism or its progeny or (sub)populations.
Endonuclease:
An enzyme that cleaves its nucleic acid substrate at internal sites in the nucleotide sequence.
Enterohepatic circulation:
Cyclical process involving intestinal re-absorption of a substance that has been excreted through bile followed by transfer back to the liver, making it available for biliary excretion again.
Epidemiology:
Study of factors determining the causes, frequency, distribution, and control of diseases in a human population.
Epigenetics:
The study of heritable changes in gene function that occur without a change in the sequence of nuclear DNA and the processes involved in the unfolding development of an organism.
Epigenetic age:
An estimate of biological age based on changes in epigenetic marks at particular locations along the genome.
Epigenetic drift:
Divergence of the epigenome as a function of age due to stochastic changes in epigenetic marks.
Epigenetic marks:
Features not directly governed by the genetic code, which include methylation of DNA and covalent modification of histone proteins. The latter may be tagged with methyl, acetyl, ubiquitin, phosphate, poly(ADP)ribose and other biochemical groups. These groups and their particular pattern of protein modification (e.g., mono-, bi-, tri-methylated at different amino acids and combinations of amino acids) modify the function of the tagged proteins and influence the way genes are expressed.
Epigenome:
The comprehensive collection of genome-wide epigenetic phenomena, including DNA-methylation patterns, chromatin modifications, and non-coding RNA.
Epigenomic reprogramming:
Resetting epigenetic marks so they resemble those of other cells from earlier developmental stages. This is of particular relevance for germline cells after the fusion of gametes when the genome is brought back into a "zero-state" of gene expression.
Epithelium:
The tissue covering the outer surface of the body, the mucous membranes and cavities of the body.
Erythema:
Reddening of the skin due to congestion of blood or increased blood flow in the skin.
Estrogen:
Sex hormone or other substance capable of developing and maintaining female characteristics of the body (note UK spelling is oestrogen).
Exogenous:
Arising outside the body.
Exposure Assessment:
Process of measuring or estimating concentration or intensity, duration and frequency of exposure to an agent. The exposure could be via the environment, consumer products or the diet, or due to occupation.
Fetotoxic:
Causing toxic, potentially lethal effects to the developing fetus.
Fibrosarcoma:
A malignant tumour arising from connective tissue (see 'tumour').
First Pass Metabolism:
rapid uptake and metabolism of an agent by the intestine or the liver, immediately after enteric absorption and before it reaches the systemic circulation.
Fluorescence In-Situ Hybridisation (FISH):
A technique that allows individual chromosomes and their centromeres to be visualised in cells.
Forestomach:
(See glandular stomach).
Free Radicals:
any molecular species capable of independent existence that contains an unpaired electron in an atomic orbital. Many radicals are unstable and highly reactive.
Full gene sequence:
the complete order of bases in a gene. This order determines which protein a gene will produce.
Gavage:
Administration of a liquid via a stomach tube, commonly used as a dosing method in toxicity studies.
Gene: The functional unit of inheritance:
a specific sequence of nucleotides along the DNA molecule, forming part of a chromosome.
Gene expression:
The process by which the information in a gene is used to create proteins or polypeptides.
Gene families:
Groups of closely related genes that make similar products.
Gene mutation:
A permanent alteration in the DNA sequence that makes up a gene, such that the sequence differs from what is found in most people. Mutations range in size; they can affect anywhere from a single DNA building block (base pair) to a large segment of a chromosome that includes multiple genes.
Gene product:
The protein or polypeptide coded for by a gene.
Genetic engineering:
Altering the genetic material of cells or organisms in order to make them capable of making new substances or performing new functions.
Genetic polymorphism:
a variation in germ-line DNA sequence among individuals, groups, or populations (e.g., a genetic polymorphism might give rise to blue eyes versus brown eyes, or population level differences in metabolic capacity). Genetic polymorphisms may be the result of chance processes or may have been induced by external agents (such as viruses or radiation). Generally, changes in DNA sequence which have been confirmed to be caused by external agents are called “mutations” rather than “polymorphisms”.
Genetic predisposition: susceptibility to a disease which is related to a polymorphism, which may or may not result in actual development of the disease.
Genetically modified organism (GMO):
An organism which has had genetic material inserted into or removed from its cells.
Genome:
All the genetic material in the chromosomes of a particular organism; its size is generally given as its total number of base pairs.
Genomic DNA:
The basic chromosome set consisting of a species-specific number of linkage groups and the genes contained therein.
Genomic imprinting:
The phenomenon whereby a small subset of all the genes in our genome are expressed according to their parent of origin.
Genomics:
The study of genes and their function.
Genotoxic:
A chemical or physical agent which has the ability to induce mutations or so-called indicator effects which are mechanistically associated with the formation of mutations (e.g., induction of DNA modifications, DNA repair, or recombination). All mutagenic substances are genotoxic but not vice versa.
Genotype:
The particular genetic pattern seen in the DNA of an individual. “Genotype” is generally used to refer to the particular pair of alleles that an individual possesses at a certain location in the genome. Compare this with phenotype.
Germ cells:
Cells that give rise to the gametes of an organism that reproduces sexually. The cells undergo mitotic and meiotic cell division in the gonads followed by cellular differentiation into mature gametes, either oocytes or sperm.
Glandular stomach:
The stomach in rodents consists of two separate regions – the forestomach and the glandular stomach. Only the glandular stomach is directly comparable to the human stomach.
Half-life:
In the context of toxicokinetics, this is the time in which the concentration of a substance in vivo will be reduced by 50%, assuming a first order elimination process.
Hazard: Set of inherent properties of a substance, mixture of substances or a process involving substances that make it capable of causing adverse effects to organisms or the environment.
Health based guidance value (HBGV):
A value indicating the amount of chemical in food that a person can consume on a regular basis usually over a lifetime without any significant risk to health.
Hepatic:
Pertaining to the liver.
Hepatocyte:
The principal cell type in the liver, possessing many metabolising enzymes (see 'metabolic activation').
Heterozygous:
having two different forms (alleles) of a gene that controls a particular characteristic, one inherited from each parent, and therefore able to pass on either form.
Histone methylation:
The modification of certain amino acids in a histone protein by the addition of methyl groups.
Histone modification:
Covalent post-translational modifications to histone proteins including methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation, which regulate gene expression. The modifications made to histones can impact gene expression by altering chromatin structure.
Histone tails:
A structural aspect of histones that are major targets for post-translational modifications of histones (see Histone modifications).
Hodgkin’s lymphoma:
Cancer of the lymphatic system.
Homeostatic:
Any self-regulation process by which biological systems tend to maintain stability while adjusting to conditions that are optimal for survival.
Horizon scanning:
The systematic examination of potential threats, opportunities and likely future developments, which are at the margins of current thinking and planning. Horizon scanning may explore novel and unexpected issues, as well as persistent problems and trends. Overall, horizon scanning is intended to improve the robustness of policies and the evidence base.
Hypoxanthine-guanine Phosphoribosyltransferase (HPRT) assay:
This assay uses cultured mammalian somatic cells to detect mutagenic agents. The principle of the method relies on the fact that mutations (caused by mutagens) destroy the functionality or the HPRT gene or protein, which is detected by using a toxic analogue. The HPRT-mutants are viable colonies that can be scored.
Hypoxanthine-guanine Phosphoribosyltransferase (HPRT) gene:
A protein coding gene. This transferase allows cells to recycle purines, a building block of DNA and RNA.
Hypermethylation:
Increase in the methylation of cytosine-guanosine base pairs in regulatory regions of DNA.
Hyperplasia:
An increase in the size of an organ or tissue due to an increase in the number of cells through cell division.
Hypertrophy:
An increase in the size of an organ or tissue due to an increase in the volume of individual cells within it.
Hypomethylation:
The loss of the methyl group in 5-methylcytosine nucleotides in DNA. Hypomethylation can be used to describe the unmethylated state of specific nucleotides or as a general phenomenon affecting large parts of the genome.
Idiosyncrasy:
Specific (and usually unexplained) reaction of an individual to e.g., a chemical exposure to which most other individuals do not react at all. General allergic reactions do not fall into this category.
In silico:
a term used to describe a computerised analysis of the structure of a chemical to assess its potential hazard.
In situ hybridisation (ISH):
Use of a DNA or RNA probe to detect the presence of the complementary DNA sequence in cloned bacterial or cultured eukaryotic cells.
In vitro:
A Latin term used to describe studies of biological material outside the living animal or plant (literally “in glass”).
In vivo:
A Latin term used to describe studies in living animals or plants (literally “in life”).
Incidence:
Number of discrete events, for example new cases of illness occurring during a given period in a specific population.
(Enzyme) Inducing agent:
A chemical which, when administered to an animal, causes an increase in the expression of a particular enzyme. For example, chlorinated dibenzodioxins are inducing agents which act via the Ah-receptor (qv) to induce P450 (qv) CYP1A1.
Intraperitoneal:
Within the abdominal cavity.
Isomer:
Isomers are two or more chemical compounds with the same molecular formula but having different properties owing to a different arrangement of atoms within the molecule.
Key event:
An empirically observable precursor step that is itself a necessary element of an AOP or MOA. A key event is a necessary, though usually not a sufficient, step in a process that results in an adverse outcome.
kilobase (kb):
A length of DNA equal to 1000 nucleotides.
Knockout animals:
Genetically engineered animals in which one or more genes, usually present and active in the normal animal, have been eliminated or inactivated.
LC50/LD50:
The concentration or dose that causes death in 50% of a group of experimental animals to which it is administered. It can be used to assess the acute toxicity of a compound but is being superseded by more refined methods.
Less than lifetime (LTL) exposure:
any exposure that is not continuous daily exposure, for example, short-term, intermediate or intermittent, or a combination of these.
Leukaemia:
A group of neoplastic disorders (see tumour) affecting blood-forming elements in the bone marrow, characterised by uncontrolled proliferation and disordered differentiation or maturation. Examples include the lymphocytic leukaemia’s which develop from lymphoid cells and the myeloid leukaemia’s which are derived from myeloid cells (producing red blood cells, mainly in bone marrow).
Ligand:
A molecule which binds to an allosteric binding site in a protein, such as a receptor.
Lipids:
Fats, substances containing a fatty acid and soluble in alcohols or ether, but insoluble in water.
Lipophilic:
'Lipid liking' - a substance which has a tendency to partition into fatty materials.
Lowest observed adverse effect level (LOAEL):
The lowest administered dose at which a statistically significant adverse effect, relative to that of the control, has been observed. Also given as LOEL when no ‘adverse’ effects are seen.
Lymphocyte:
A type of white blood cell that plays central roles in adaptive immune responses.
Lymphoma:
Malignant tumours arising from lymphoid tissues. They are usually multifocal, involving lymph nodes, spleen, thymus and sometimes bone marrow, and other sites outside the anatomically defined lymphoid system. (See also 'tumour').
Malformations:
The inheritance of an abnormal or anomalous formation of tissues and organs often referred to as a deformity.
Malignant tumour (synonym:
cancer): A tumour (qv) composed of increasingly abnormal cells in term of their form and function. Some well differentiated examples still retain characteristics of their tissues of origin but these are progressively lost in moderately and poorly differentiated malignancies. Most malignant tumours grow rapidly, spread progressively through adjacent tissues and metastasise to distant sites.
Margin of exposure (MOE) approach:
A methodology that allows the comparison of the risks posed by substances when it is not possible or not appropriate to establish a HBGV. This would include substances that are genotoxic and carcinogenic, and contaminants for which there is insufficient information to establish a Tolerable Daily Intake The MOE approach uses a reference point (or POD), often taken from an animal study, corresponding to a dose that causes no or a low response (for example the NAOEL, LOAEL, BMDL10). This reference point is then compared with various exposure estimates in humans. The lower the MOE, the greater the concern. The MOE considered to be of low or negligible concern is context specific. In general, for substances that are genotoxic and carcinogenic, and MOE of >10,000, when based on a reference point from an animal study, would be considered of low concern. For a non-genotoxic, non-carcinogenic contaminant, an MOE of > 100 would be considered of negligible concern.
Margin of safety (MOS) approach:
A methodology used to assess relative risk when there is exceedance of a HBGV. The MOS is expressed as the ratio of the HBGV to measured or estimated exposure. The lower the MOS is below 1, the greater the concern.
Maximum tolerated dose:
The MTD for a long-term study of carcinogenicity is a dose that produces minimal signs of toxicity on repeated administration, meaning no more than a 10% weight decrement, as compared to the appropriate control groups; and does not produce mortality, clinical signs of toxicity, or pathologic lesions (other than those that may be related to a neoplastic response) that would be predicted to shorten the animal's natural life span.
Mechanism of action:
an understanding of the molecular basis for an effect and its detailed description, so causation can be established in molecular terms.
Meiosis:
The process of cell division in sexually reproducing organisms that reduces the number of chromosomes in reproductive cells from diploid to haploid leading to the production of gametes in animals and spores in plants. During the first meiotic division there is homologue pairing, efficient intergenic recombination between homologues during pairing, and the suppression of sister chromatid separation. S phase is absent at the start of the second meiotic division. Thus, the outcome of meiosis should be four genetically unique haploid cells.
Messenger RNA (mRNA):
The DNA of a gene is transcribed (see transcription) into mRNA molecules, which then serve as a template for the synthesis of proteins.
Meta-analysis:
In the context of epidemiology, a statistical analysis of the results from independent studies, which aims to produce a single estimate of an effect.
Metabolic activation:
Metabolism of a compound leading to an increase in its activity, whether beneficial (e.g., activation of a pro-drug) or deleterious (e.g., activation to a toxic metabolite).
Metabolic activation system:
A cell-free preparation (e.g., from the livers of rats pre-treated with an inducing agent (qv)) added to in vitro tests to mimic the metabolic activation typical of mammals.
Metabolism:
Chemical modification of a compound by enzymes within the body, for example by reactions such as hydroxylation (see P450), epoxidation or conjugation. Metabolism may result in activation, inactivation, change in activity, accumulation or excretion of the compound.
Metabolite:
Product formed by metabolism of a compound.
Metabolomics:
The measurement of the amounts (concentrations) and locations of all metabolites in a cell.
Metabonomics:
Metabonomics is a subset of metabolomics and is defined as the quantitative measurement of the multiparametric metabolic responses of living systems to pathophysiological stimuli or genetic modification.
Metaphase:
Stage of cell division (mitosis and meiosis) during which the chromosomes are arranged on the equator of the nuclear spindle (the collection of microtubule filaments which are responsible for the movement of chromosomes during cell division). As the chromosomes are most easily examined in metaphase, cells are arrested at this stage for microscopical examination for chromosomal aberrations (qv) - known as metaphase analysis.
Metastasis:
The process whereby malignant cells become detached from the primary tumour mass, disseminate (mainly in the blood stream or in lymph vessels) and seed out in distant sites where they form secondary or metastatic tumours. Such tumours tend to develop at specific sites and their anatomical distribution is often characteristic, i.e., it is non-random.
Microbiome (Human):
Human microbiome is the full array of microorganisms (the microbiota) that live on and in humans and, more specifically, the collection of microbial genomes that contribute to the broader genetic portrait, or metagenome, of a human. Often a subset of the microbiome is the subject of interest, for example the intestinal or dermal microbiome.
Micronuclei:
Whole or fragmented chromosomes that fail to segregate normally during cell division and may be lost from the main nuclei but remain in the body of the cell forming micronuclei. Centromere positive micronuclei contain DNA and/or protein material derived from the centromere. The presence of centromere positive micronuclei following exposure to chemicals in vitro or in vivo can be used to evaluate the aneugenic potential of chemicals.
Minimal risk level:
defined in this document as an estimate of daily human exposure to a chemical, identified by expert judgement, that is likely to be associated with a negligible risk of carcinogenic effect over a specified duration of exposure (usually a lifetime).
Mitogen:
A stimulus which provokes cell division in somatic cells.
Mitosis:
The process in cell division in somatic cells by which the nucleus divides, typically consisting of four stages, prophase, metaphase, anaphase, and telophase, and normally resulting in two new nuclei, each of which contains a complete copy of the parental chromosomes. The outcome of mitosis should be two genetically identical diploid cells.
Mode of Action:
a biologically plausible sequence of key events leading to an observed effect supported by robust experimental observations and mechanistic data. It describes key cytological and biochemical events, i.e., those that are both measurable and necessary to the observed outcome, in a logical framework. It contrasts with mechanism of action.
Mode of genotoxic action (MoGA):
The mode of action of a genotoxicant refers to the underlying events involved in the process whereby the chemical induces genotoxic effects. In order for a specific mode of action to be supported there needs to be evidence from robust mechanistic data to establish a biologically plausible explanation. Mode of genotoxic action should be distinguished from the term mechanism of action. The latter relates to having sufficient understanding of the molecular basis of the chemical genotoxicity to establish causality. Thus, mechanism of action is at the other end of a continuum from little or no evidence of mode of genotoxic action to scientific proof of mechanism of action.
Molecular initiating event (MIE):
the initial point of chemical/stressor interaction at the molecular level within the organism that results in a perturbation that starts the AOP.
Mouse lymphoma assay:
An in vitro assay for gene mutation in mammalian cells using a mouse lymphoma cell line L5178Y, which is heterozygous for the gene (carries only one functional allele rather than a pair) for the enzyme thymidine kinase (TK+/-). Mutation of that single gene is measured by resistance to toxic trifluorothymidine. Mutant cells produce two forms of colony - large, which represent mutations within the gene and small, which represent large genetic changes in the chromosome such as chromosome aberrations. Thus, this assay can provide additional information about the type of mutation which has occurred if colony size is scored.
Mucosal:
Regarding the mucosa or mucous membranes, consisting of epithelium containing glands secreting mucus, with underlying layers of connective tissue and muscle.
Multigenerational effects:
Effect seen in exposed generations, including those that may have been exposed in utero, as offspring or gametes. For effects in unexposed generations see ‘Transgenerational effects’.
Murine:
Often taken to mean “of the mouse”, but strictly speaking means of the Family Muridae which includes rats and squirrels.
Mutagen:
is a physical or chemical agent that changes the genetic information (usually DNA) of an organism that can be inherited by daughter cells.
Mutation:
A permanent change in the amount or structure of the genetic material in an organism or cell, which can result in a change in phenotypic characteristics. The alteration may involve a single gene, a block of genes, or a whole chromosome. Mutations involving single genes may be a consequence of effects on single DNA bases (point mutations) or of large changes, including deletions, within the gene. Changes involving whole chromosomes may be numerical or structural. A mutation in the germ cells of sexually reproducing organisms may be transmitted to the offspring, whereas a mutation that occurs in somatic cells may be transferred only to descendent daughter cells.
Mutational signatures:
Mutational signatures are characteristic profiles of mutation types arising from specific mutagenesis processes such as DNA replication infidelity, exogenous and endogenous genotoxins exposures, defective DNA repair pathways and DNA enzymatic editing.
Mycotoxin:
Toxic compound produced by a fungus.
Nanomaterial:
A natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1 nm - 100 nm.
Neoplasia:
the abnormal proliferation of benign or malignant cells.
Neoplasm:
See 'tumour'.
Neoplastic:
Abnormal cells, the growth of which is more rapid that that of other cells of the same tissue type.
Neural tube defect (NTD):
Is a birth defect in which an opening in the spine or cranium remains from early in human development.
Neurobehavioural:
Of behaviour determined by the nervous system.
Neurotransmitter:
A chemical that is released from a nerve cell which thereby transmits an impulse from a nerve cell to another nerve, muscle, organ, or other tissue. A neurotransmitter is a messenger of neurologic information from one cell to another.
No observed adverse effect level (NOAEL):
The highest administered dose at which no statistically significant adverse effect has been observed in comparison to the control. Also given as NOEL when no ‘adverse’ effects are seen.
Non-Hodgkin lymphomas:
(NHLs) are a diverse group of hematologic cancers which encompass any lymphoma other than Hodgkin’s Lymphoma.
No observed genotoxic effect level (NOGEL):
This is the highest experimental dose level where no statistically significant increase in the genotoxic effect measured in the study is identified.
Nucleic acid:
One of the family of molecules which includes the DNA and RNA molecules. Nucleic acids were so named because they were originally discovered within the nucleus of cells, but they have since been found to exist outside the nucleus as well.
Nucleosome:
A repeating subunit of DNA packaging consisting of DNA wound in sequence around histone proteins.
Nucleotide:
the "building block" of nucleic acids, such as the DNA molecule. A nucleotide consists of a nucleoside attached a phosphate group. A nucleoside comprises one of four bases - adenine, guanine, cytosine, or thymine - attached to a sugar group. In DNA the sugar group is deoxyribose, while in RNA (a DNA-related molecule which helps to translate genetic information into proteins), the sugar group is ribose, and the base uracil substitutes for thymine. Each group of three nucleotides in a gene is known as a codon. A nucleic acid is a long chain of nucleotides joined together, and therefore is sometimes referred to as a "polynucleotide."
Null allele:
Mutations that result in absence of gene product or a non-functional product.
Null hypothesis:
type of conjecture used in statistical tests, which are formal methods of reaching conclusions or making decisions on the basis of data. In toxicology, a common null hypothesis is that there is no effect of treatment with a substance. Statistical testing may enable a conclusion that this is most likely incorrect, i.e., the null hypothesis is rejected with a stated probability of error, or it is not possible to reach a conclusion. It is not possible by conventional statistical testing to prove the null hypothesis is most likely correct, i.e., that there is no effect. This is the axiomatic difficulty of “proving a negative”.
Odds ratio (OR):
The odds of disease in an exposed group divided by the odds of disease in an unexposed group.
Oedema:
Excessive accumulation of fluid in body tissues.
Oligonucleotide:
A molecule made up of a small number of nucleotides, typically fewer than 25.
‘Omics’ technologies:
A scientific subdiscipline that combines the technologies of genomics and bioinformatics to identify and characterise mechanisms of action of known and suspected toxicants. The collective term ‘omics’ refers to the genomic (DNA sequence analysis) and post-genomic (e.g., transcriptomics, proteomics, metabolomics, epigenomics) technologies that are used for the characterisation and quantitation of pools of biological molecules (e.g. DNA, mRNAs, proteins, metabolites), and the exploration of their roles, relationships and actions within an organism.
Oncogene:
A gene which is associated with the development of cancer (see proto-oncogene).
Pharmacodynamics:
The process of interaction of drugs with target sites and the subsequent reactions leading to the desired biological effects (see toxicodynamics).
Pharmacokinetics:
Description of the fate of drugs in the body, including a mathematical account of their absorption, distribution, metabolism and excretion (see toxicokinetics).
Pharmacogenomics:
The science of understanding the correlation between an individual patient's genetic make-up (genotype) and their response to drug treatment. Some drugs work well in some patient populations and not as well in others. Studying the genetic basis of patient response to therapeutics allows drug developers and medical practitioners to design and use therapeutic treatments more effectively.
Phenotype:
The observable physical, biochemical and physiological characteristics of a cell, tissue, organ or individual, as determined by its genotype and the environment in which it develops.
Phenotypic change:
A change in the observable physical or biochemical characteristics of an organism, as determined by both genetic makeup and environmental influences.
Physiologically based pharmacokinetic (PBPK) model:
A mathematical model which is used to predict the absorption, distribution, metabolism and excretion of a chemical substance in humans.
Phytoestrogen:
Any plant substance or metabolite that can mimic or modulate the actions of endogenous oestrogens, usually by binding to oestrogen receptors, and which can therefore induce biological responses.
Pig-A gene mutation assay:
An assay which utilises the Pig-A gene which codes for one subunit of a glycosylphosphatidyl inositol anchor protein. Loss of function arising from Pig-A mutations can readily be assessed using straightforward immunochemistry and flow cytometric methods, thus making it useful to measure gene mutations induced by chemicals or radiation.
Plasmid:
A structure composed of DNA that is separate from the cell's genome (qv). In bacteria, plasmids confer a variety of traits and can be exchanged between individuals, even those of different species. Plasmids can be constructed and manipulated in the laboratory to deliver specific genetic sequences into a cell.
Point of departure:
a dose associated with a defined level of effect, which can be determined empirically or by modelling dose-response data from experimental studies, from which a health-based guidance value can be established, or which can be used for a margin of exposure assessment. Examples include a BMDL, NOAEL or LOAEL.
Polymer:
A very large molecule comprising a chain of many similar or identical molecular sub units (monomers) joined together (polymerised). An example is the polymer glycogen, formed from linked molecules of the monomer glucose.
Polymerase chain reaction (PCR):
A method for creating millions of copies of a particular segment of DNA. PCR can be used to amplify the amount of a particular DNA sequence until there are enough copies available to be detected.
Polymorphism:
(see genetic polymorphism)
32P postlabelling assay:
An experimental method designed to measure low levels of DNA adducts induced by chemical treatment. It involves labelling of adducted nucelosides from digested DNA with 32P and their quantification following chromatographic separation.
Prevalence:
The number of discrete cases, for example of a disease, that are present in a population at a given time.
Primer:
Short pre-existing polynucleotide chain to which new deoxyribonucleotides can be added by DNA polymerase.
Primordial germ cells:
Highly specialised cells that are precursors of gametes, which, following meiosis, develop as haploid sperm and eggs that generate a new organism upon fertilisation.
Proteomics:
The analysis of the entire protein complement of a cell, tissue, or organism under a specific, defined set of conditions.
Proto-oncogene:
One of a group of normal genes that are concerned with the control of cellular proliferation and differentiation. They can be activated in various ways to forms (oncogenes) which are closely associated with one or more steps in carcinogenesis. Activating agents include chemicals and viruses. The process of proto-oncogene activation is thought to play an important part at several stages in the development of tumours.
Quantal Data:
When the response for an individual unit (well, animal etc) is a binary value, such as alive / dead, or response / no response, the data are treated as quantal. The responses are assumed to follow a binomial distribution within each dose group. This assumption is required for the calculations of confidence intervals and the p values resulting from statistical tests.
ras oncogene:
The Ras protein family are a class of protein called small GTPase and have important roles in cell signalling. The ras gene is the most common oncogene involved in human cancer - mutations that permanently activate ras are found in 20-25% of all human tumours and up to 90% in certain types of cancer (e.g., pancreatic cancer).
Receptor:
A small, discrete protein in the cell membrane or within the cell with which specific molecules interact to initiate a change in the working of a cell.
Recombinant DNA:
DNA molecules that have been created by combining DNA from more than one source.
Reference nutrient intake (RNI):
An amount of the nutrient that is sufficient, or more than sufficient, to ensure adequate nutrient function for most (usually at least 97%) people in a group. If the average intake of a group is at the RNI, then the risk of deficiency in the group is very small.
Regulatory gene:
A gene which controls the protein-synthesising activity of other genes.
Relative potency factor (RPF):
The toxic potency of a substance expressed relative to that of an index chemical to enable cumulative risk assessment (qv). The RPF is similar to the TEF (qv) but is used when the information on common MIEs, toxicokinetics and outcomes of the members of an assessment group is less reliable than that required for application of the TEF approach.
Relative risk:
A measure of the association between exposure and outcome. The rate of disease in the exposed population divided by the rate of disease among the unexposed population in a cohort study or a population-based case control study. A relative risk of 2 means that the exposed group has twice the disease risk compared to the unexposed group.
Reporter gene:
A gene that encodes an easily assayed product that is coupled to the upstream sequence of another gene and transfected (qv) into cells. The reporter gene can then be used to see which factors activate response elements in the upstream region of the gene of interest.
Risk:
Probability that a harmful event (death, injury or loss) arising from exposure to a chemical or physical agent may occur under specific conditions.
Risk assessment:
process of evaluating a potential hazard, likelihood of suffering, or any adverse effects from certain human activities. Comprised of the four aspects, hazard identification, hazard characterisation, exposure assessment and risk characterisation. Can be carried out retrospectively or prospectively.
Risk management:
process designed to identify, contain, reduce, or eliminate the potential for harm to the human population; usually concerned with the delivery system and site rather than performance.
Ribonucleic acid (RNA):
a molecule similar to DNA, in that it is a nucleic acid comprised of a chain of nucleotides. However, unlike DNA, RNA exists as a single-stranded chain. RNA has various biological roles in coding, decoding, regulation and expression of genes.
Sarcoma:
cancer that arises from transformed cells of mesenchymal (connective tissue) origin.
Serotonergic:
Denoting a nerve ending that releases and or stimulated by serotonin.
Signal induction pathway:
The molecular pathways that signal (i.e., turn on or off) biochemical pathways or biological functions (e.g., biochemical pathways leading to nerve conduction).
Single nucleotide polymorphism (SNP):
DNA sequence variations that occur when a single nucleotide in the genome sequence is altered. For example, a SNP might change the DNA sequence AAGGCTAA to ATGGCTAA.
Strand breaks:
Relating to DNA, a single strand break occurs when there is a break in double-stranded DNA in which only one of the two strands has been cleaved; the two strands have not separated from each other. Double strand breaks occur when both strands in the double helix are severed and are particularly hazardous to the cell because they can lead to genome rearrangements.
Sister chromatid exchange (SCE):
Exchange of genetic material between two subunits of a replicated chromosome.
Somatic cells:
Any biological cell that forms part of the body of an organism, excluding reproductive cells and undifferentiated stem cells.
Stakeholder:
A person or organisation representing the interests and opinions of a group with an interest in the outcome of (for example) a review or policy decision.
Statistical significance:
a conclusion drawn when, after carrying out a statistical test of the null hypothesis of no effect, the hypothesis is considered unlikely to be true. The criterion for the decision is often a probability (p) value, chosen to be, but not necessarily, p<0.05.
Stem cell:
an unspecialized cell capable of perpetuating itself through cell division and having the potential to give rise to differentiated cells with specialized functions.
Suppressor gene:
A gene which helps to reverse the effects of damage to an individual's genetic material, typically these are effects which might lead to uncontrolled cell growth (as would occur in cancer). A suppressor gene may, for example, code for a protein which checks genes for misspellings, and/or which triggers a cell's self-destruction if too much DNA damage has occurred.
Systematic review:
A formalised review that has been prepared using a documented systematic approach to minimising biases and random errors.
Systems biology:
The computational and mathematical analysis and modelling of complex biological systems.
Systems toxicology:
The integration of classical toxicology with quantitative analysis of large networks of molecular and functional changes occurring across multiple levels of biological organisation.
T25:
the dose eliciting a 25% increase in the incidence of a specific tumour above the background level.
TD50:
For any particular sex, strain, species and set of experimental conditions, the TD50 is the dose rate (in mg/kg body weight/day) that, if administered chronically for a standard period - the "standard lifespan" of the species-will halve the mortality-corrected estimate of the probability of remaining tumourless throughout that period.
Teratogen:
A substance that can cause congenital malformations (structural defects) in a developing fetus following maternal exposure.
Testicular dysgenesis syndrome (TDS):
The hypothesis that maldevelopment (dysgenesis) of the fetal testis results from hormonal or other malfunctions of the testicular somatic cells which in turn predispose a male to the disorders that comprise the TDS, i.e., congenital malformations (cryptorchidism and hypospadias) in babies and testis cancer and low sperm counts in young men.
Threshold:
the level of dose or exposure below which there is no effect above that in the control group or population. There are several different uses of the term threshold, for example observable threshold, biological threshold, population threshold.
Threshold of toxicological concern (TTC):
a pragmatic, scientifically valid methodology to prioritise substances of unknown toxicity found in food for further evaluation. It is used when there are limited chemical-specific toxicity data and can be used for substances with or without structural alerts for genotoxicity and for cancer and non-cancer endpoints.
Tolerable daily intake (TDI):
An estimate of the amount of contaminant, expressed on a body weight basis (e.g., mg/kg bodyweight), that can be ingested daily over a lifetime without appreciable health risk. The term is preferred for substances that are unintentionally present.
Tolerable upper level (TUL):
The highest level of nutrient that is likely to pose no risk of adverse health effects for almost all individuals in the general population. As intake increases above the TUL, the risk of adverse effects increases.
Toxic equivalency factor (TEF):
A measure of relative toxicological potency of a chemical compared to a well characterised reference compound. TEFs can be used to sum the toxicological potency of a mixture of chemicals which are all members of the same chemical class, having common structural, toxicological and biochemical properties. TEF systems have been published for the chlorinated dibenzodioxins, dibenzofurans and dioxin-like polychlorinated biphenyls, and for polycyclic aromatic hydrocarbons.
Total toxic equivalent (TEQ):
Is a method of comparing the total relative toxicological potency within a sample. It is calculated as the sum of the products of the concentration of each congener multiplied by the toxic equivalency factor (TEF).
Toxicodynamics:
The process of interaction of chemical substances with target sites and the subsequent reactions leading to adverse effects.
Toxicogenic:
producing or capable of producing toxins, e.g., a fungal strain.
Toxicokinetics:
The description of the fate of potentially toxic chemicals in the body, including a mathematical account of their absorption, distribution, metabolism and excretion, particularly at doses that are toxic. (see pharmacokinetics)
Transcription:
the process during which the information in a piece of DNA (qv) is used to construct an mRNA (qv) molecule.
Transcriptomics:
Techniques used to identify mRNA from actively transcribed genes.
Transgenerational effects:
Effects seen in generations that have not been exposed, either directly to the substance under consideration or indirectly as offspring or gametes via parental exposure. For effects in exposed populations, see ‘multigenerational effects’.
Transfer RNA (tRNA):
RNA molecules which bond with amino acids and transfer them to ribosomes, where protein synthesis is completed.
Transfection:
A process by which exogenous genetic material (DNA or RNA) is introduced into a cell with the object of altering the phenotype or genotype of the cell.
Transgenic:
Genetically modified to contain genetic material from another species (see also genetically modified organism).
Transgenic animal models:
Animals which have extra (exogenous) fragments of DNA incorporated into their genomes. This may include reporter genes to assess in-vivo effects such as mutagenicity in transgenic mice containing a recoverable bacterial gene (lacZ or lac I). Other transgenic animals may have alterations of specific genes believed to be involved in disease processes (e.g., cancer). For example, strains of mice have been bred which carry an inactivated copy of the p53 tumour suppressor gene, or an activated form of the ras oncogene which may enhance their susceptibility of the mice to certain types of carcinogenic chemicals.
Translation:
In molecular biology, the process during which the information in mRNA molecules is used to construct proteins.
Tumour (Synonym - neoplasm):
A mass of abnormal, disorganised cells, arising from pre-existing tissue, which are characterised by excessive and uncoordinated proliferation and by abnormal differentiation. Benign tumours show a close morphological resemblance to their tissue of origin; grow in a slow expansile fashion; and form circumscribed and (usually) encapsulated masses. They may stop growing and they may regress. Benign tumours do not infiltrate through local tissues, and they do not metastasise (qv). They are rarely fatal. Malignant tumours (synonym - cancer) resemble their parent tissues less closely and are composed of increasingly abnormal cells in terms of their form and function. Well differentiated examples still retain recognisable features of their tissue of origin, but these characteristics are progressively lost in moderately and poorly differentiated malignancies: undifferentiated or anaplastic tumours are composed of cells which resemble no known normal tissue. Most malignant tumours grow rapidly, spread progressively through adjacent tissues and metastasise to distant sites. Tumours are conventionally classified according to the anatomical site of the primary tumour and its microscopical appearance, rather than by cause. Benign tumours may evolve to the corresponding malignant tumours; examples involve the adenoma → carcinoma sequence in the large bowel in humans, and the papilloma → carcinoma sequence in mouse skin.
Tumour initiation:
A term originally used to describe and explain observations made in laboratory models of multistage carcinogenesis, principally involving repeated applications of chemicals to the skin of mice. Initiation, in such contexts, was the first step whereby small numbers of cells were irreversibly changed or initiated. Subsequent, separate events (see tumour promotion) resulted in the development of tumours. It is now recognised that these early, irreversible heritable changes in initiated cells were due to genotoxic damage, usually in the form of somatic mutations and the initiators used in these experimental models can be regarded as genotoxic carcinogens.
Tumour microenvironment:
This is a complex system of many cell types, including cancer cells, fibroblasts, endothelial cells, leukocytes and antigen-presenting cells, together with connective tissue. The microenvironment is integral in determining the functionality, physiology and spread (metastasis) of cancer.
Tumour promotion:
Originally used, like ‘tumour initiation’ to describe events in multistage carcinogenesis in experimental animals. In that context, promotion is regarded as the protracted process whereby initiated cells undergo clonal expansion to form overt tumours. The mechanisms of clonal expansion are diverse, but include direct stimulation of cell proliferation, repeated cycles of cell damage and cell regeneration and release of cells from normal growth-controlling mechanisms. Initiating and promoting agents were originally regarded as separate categories, but the distinction between them is becoming increasingly hard to sustain. The various modes of promotion are non-genotoxic, but it is incorrect to conclude that ‘non-genotoxic carcinogen’ and ‘promoter’ are synonymous.
Uncertainty factor:
Value used in extrapolation from a reference point (or POD), determined in experimental animals, to humans (assuming that humans may be more sensitive) or from a sub-population of individuals to the general population: for example, a value applied to the NOAEL to establish an ADI or TDI. The value depends on the size and type of population to be protected and the quality of the toxicological information available.
Unscheduled DNA synthesis (UDS):
DNA synthesis that occurs at some stage in the cell cycle other than the S period (the normal or 'scheduled' DNA synthesis period), in response to DNA damage. It is usually associated with DNA repair.
Volume of distribution:
Apparent volume of fluid required to contain the total amount of a substance in the body at the same concentration as that present in the plasma, assuming equilibrium has been attained.
Weight of evidence:
This approach uses a combination of several independent sources of evidence (e.g., toxicological or genotoxicity data) to arrive at a conclusion regarding potential hazard (such as mutagenicity).
WHO-TEQs:
The system of Toxic Equivalency Factors (TEFs) used in the UK and a number of other countries to express the concentrations of the less toxic dioxin-like compounds (16 PCDDs/PCDFs and 12 PCBs) as a concentration equivalent to the most toxic dioxin 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is that set by the World Health Organisation (WHO), and the resulting overall concentrations are referred to as WHO-TEQs (Total toxic equivalents) (see also Toxic Equivalency Factor).
Xenobiotic:
A chemical foreign to the biologic system.
Xenoandrogen:
A ‘foreign’ compound with androgenic activity (see androgen).
Xenoestrogen:
A 'foreign' compound with oestrogenic activity (see oestrogen).
Organisational abbreviations
COC: Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment:
is an independent scientific committee that provides advice the government and government agencies on whether substances are likely to cause cancer.
COM: Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment:
is an independent scientific committee that assesses and advises the government and government agencies on mutagenic risks to humans.
COT Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment:
is an independent scientific committee that provides advice to the government and government agencies on matters concerning the toxicity of chemicals.
EFSA European Food Safety Authority
Expert Group on Vitamins and Minerals (EVM):
An independent UK expert advisory committee which was asked to advise on safe levels of intakes of vitamins and minerals in food supplements and fortified foods.
Annex 7 – Previous Publications
In this guide
In this guidePublications produced by the Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment
1991 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. HMSO ISBN 0 11 321529 0 Price £9.50.
1992 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. HMSO ISBN 0 11 321604-1 Price £11.70.
1993 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. HMSO ISBN 0 11 321808-7 Price £11.95.
1994 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. HMSO ISBN 0 11 321912-1 Price £12.50.
1995 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. HMSO ISBN 0 11 321988-1 Price £18.50.
1996 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. The Stationery Office ISBN 0 11 322115-0 Price £19.50.
1997 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Department of Health.*
1998 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Department of Health.*
1999 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Department of Health.*
2000 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Department of Health.*
2001 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/0681/0802.++
2002 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/0838/0803.++
2003 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/0900/0504.++
2004 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/0992/0804.++
2005 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/1098/0906.++
2006 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/1184/0707.++
2007 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/1260/0608.++
2008 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, FSA/1410/0709.++
2009 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, July 2010.++
2010 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, June 2011.++
2011 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, July 2012.
2012 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, April 2014.
2013 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, March 2015.
2014 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, November 2015.
2015 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, July 2016.
2016 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, August 2017.
2017 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health, October 2017.
2018 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health and Social Care, November 2018.
2019 Annual Report of Committees on Toxicity, Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Food Standards Agency/Department of Health and Social Care, December 2019.
Guidelines for the Testing of Chemicals for Toxicity DHSS Report on Health and Social Subjects 27 HMSO ISBN 0 11 320815 4 Price £4.30.
Guidelines for the Evaluation of Chemicals for Carcinogenicity DH Report on Health and Social Subjects 42 HMSO ISBN 0 11 321453 7 Price £7.30.
Guidelines for the Testing of Chemicals for Mutagenicity DH Report on Health and Social Subjects 35 HMSO ISBN 0 11 321222 4 Price £6.80.
Guidelines for the Preparation of Summaries of Data on Chemicals in Food, Consumer Products and the Environment submitted to DHSS Report on Health and Social Subjects 30 HMSO ISBN 0 11 321063 9 Price £2.70.
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Peanut Allergy, Department of Health (1998).**
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Organophosphates, Department of Health (1998).**
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Adverse Reactions to Food and Food Ingredients, Food Standards Agency (2000).++
Guidance on a Strategy for testing of chemicals for Mutagenicity. Department of Health (2000).*
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Risk Assessment of Mixtures of Pesticides and Similar Substances, Food Standards Agency, FSA/0691/0902 (2002).++
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Phytoestrogens and Health, Food Standards Agency, FSA/0826/0503 (2002).++
Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment: Variability and Uncertainty in Toxicology of Chemicals in Food, Consumer Products and the Environment, FSA/1150/0307 (2007).++
Guidance on a Strategy for the Risk Assessment of Chemical Carcinogens. Department of Health (2004).+
*Available at: Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment - GOV.UK (www.gov.uk).
** Available at: Le blog de l'actu européenne - EuropArchive.org.
+ Available at: Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment - GOV.UK.
++ Available at: All COT Reports.
Report of the Synthesising Epidemiological Evidence Subgroup (SEES) of the Committee on Toxicity and Committee on Carcinogenicity, Food Standards Agency/Department of Health and Social Care, September 2018. COT joint reports | Committee on Toxicity.
Available at: [ARCHIVED CONTENT] Synthesising Epidemiology Evidence Subgroup (SEES) Report | Food Standards Agency (nationalarchives.gov.uk).
Joint COT and COC Synthesis and Integration of Epidemiological and Toxicological Evidence subgroup (SETE). Food Standards Agency/Department of Health and Social Care, November 2021. SETE | Committee on Toxicity (food.gov.uk).